
Genomic Signatures of SARS-CoV-2 Associated with Patient Mortality




Abstract
:1. Background
2. Methods
2.1. Viral Sequence Data and Associated Patient Metadata
2.2. Data Analysis
2.3. Analysis of Validation Dataset
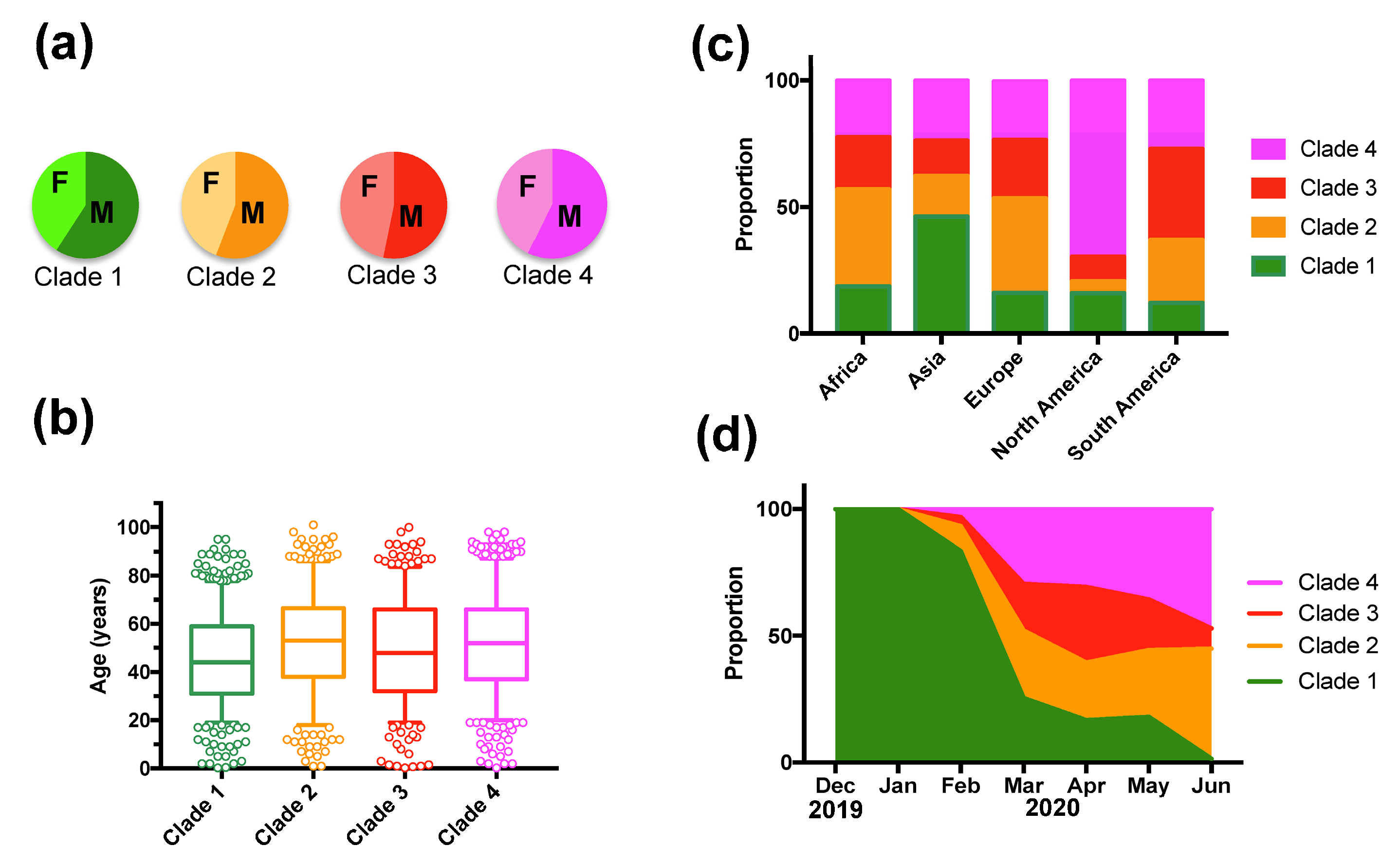
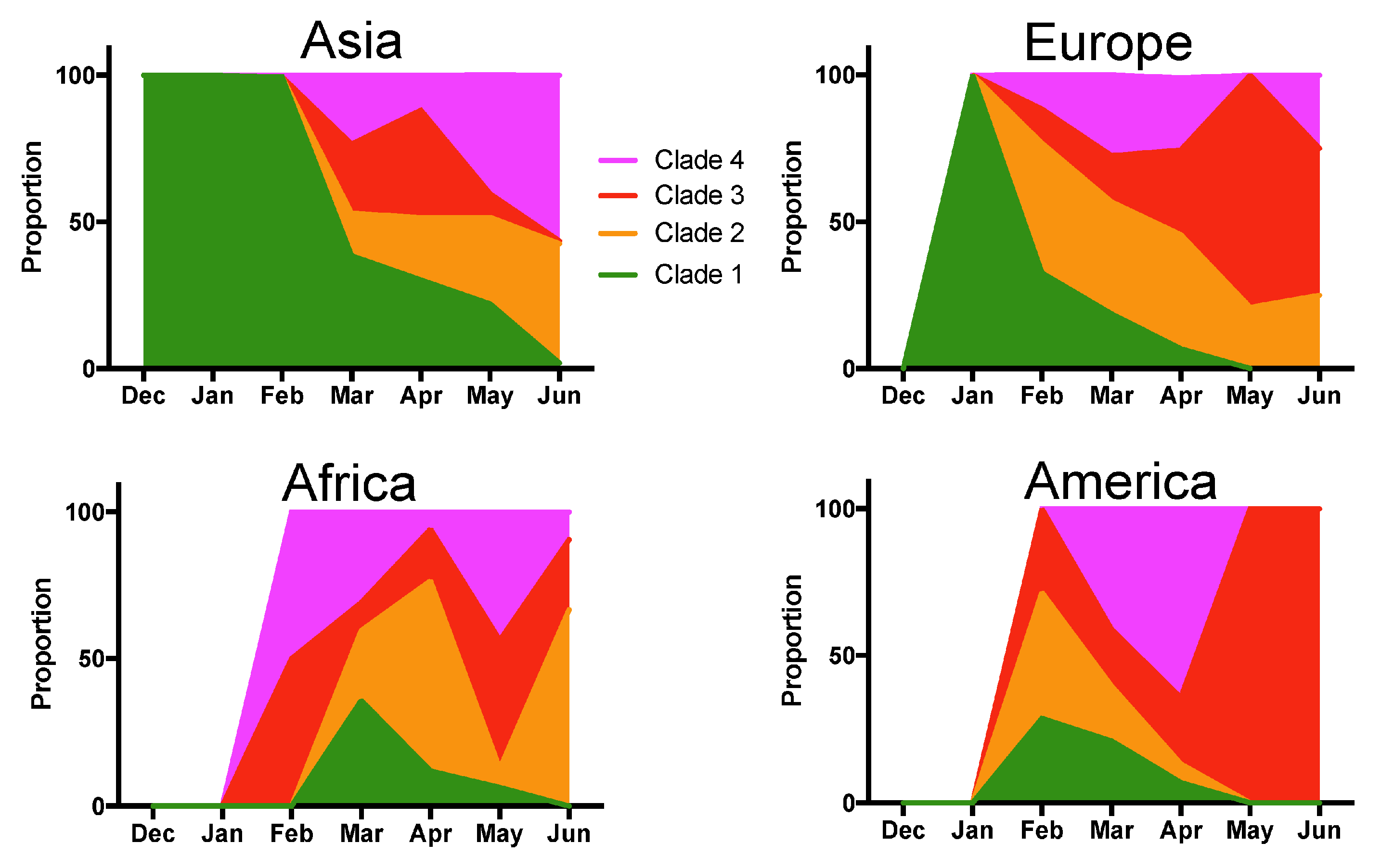
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Johns Hopkins University, Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu/ (accessed on 28 October 2020).
- Koutsakos, M.; Kedzierska, K. A race to determine what drives COVID-19 severity. Nature 2020, 582, 366–368. [Google Scholar] [CrossRef] [PubMed]
- Team, C.C.-R. Geographic Differences in COVID-19 Cases, Deaths, and Incidence-United States, February 12-April 7, 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 465–471. [Google Scholar] [CrossRef] [Green Version]
- Docherty, A.B.; Harrison, E.M.; Green, C.A.; Hardwick, H.E.; Pius, R.; Norman, L.; Holden, K.A.; Read, J.M.; Dondelinger, F.; Carson, G.; et al. Features of 20 133 UK patients in hospital with covid-19 using the ISARIC WHO Clinical Characterisation Protocol: Prospective observational cohort study. BMJ 2020, 369, m1985. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Hayek, S.S.; Wang, W.; Chan, L.; Mathews, K.S.; Melamed, M.L.; Brenner, S.K.; Leonberg-Yoo, A.; Schenck, E.J.; Radbel, J.; et al. Factors Associated With Death in Critically Ill Patients With Coronavirus Disease 2019 in the US. JAMA Intern. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tan, Y.; Ling, Y.; Lu, G.; Liu, F.; Yi, Z.; Jia, X.; Wu, M.; Shi, B.; Xu, S.; et al. Viral and host factors related to the clinical outcome of COVID-19. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- Hadjadj, J.; Yatim, N.; Barnabei, L.; Corneau, A.; Boussier, J.; Smith, N.; Pere, H.; Charbit, B.; Bondet, V.; Chenevier-Gobeaux, C.; et al. Impaired type I interferon activity and inflammatory responses in severe COVID-19 patients. Science 2020. [Google Scholar] [CrossRef] [PubMed]
- Gussow, A.B.; Auslander, N.; Faure, G.; Wolf, Y.I.; Zhang, F.; Koonin, E.V. Genomic determinants of pathogenicity in SARS-CoV-2 and other human coronaviruses. Proc. Natl. Acad. Sci. USA 2020, 117, 15193–15199. [Google Scholar] [CrossRef] [PubMed]
- Pachetti, M.; Marini, B.; Benedetti, F.; Giudici, F.; Mauro, E.; Storici, P.; Masciovecchio, C.; Angeletti, S.; Ciccozzi, M.; Gallo, R.C.; et al. Emerging SARS-CoV-2 mutation hot spots include a novel RNA-dependent-RNA polymerase variant. J. Transl. Med. 2020, 18, 179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumonteil, E.; Herrera, C. Polymorphism and selection pressure of SARS-CoV-2 vaccine and diagnostic antigens: Implications for immune evasion and serologic diagnostic performance. Pathogens 2020, 9, 584. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Foley, B.; Giorgi, E.E.; Bhattacharya, T.; Parker, M.D.; et al. Spike mutation pipeline reveals the emergence of a more transmissible form of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Ozono, S.; Zhang, Y.; Ode, H.; Seng Tan, T.; Imai, K.; Miyoshi, K.; Kishigami, S.; Ueno, T.; Iwatani, Y.; Suzuki, T.; et al. Naturally mutated spike proteins of SARS-CoV-2 variants show differential levels of cell entry. bioRxiv 2020. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Wongtrakoongate, P.; Thawornwattana, Y.; Hongeng, S.; Thitithanyanont, A. SARS-CoV-2 genetic variations associated with COVID-19 severity. medRxiv 2020. [Google Scholar] [CrossRef]
- Lorenzo-Redondo, R.; Nam, H.H.; Roberts, S.C.; Simons, L.M.; Jennings, L.J.; Qi, C.; Achenbach, C.J.; Hauser, A.R.; Ison, M.G.; Hultquist, J.F.; et al. A Unique Clade of SARS-CoV-2 Viruses is Associated with Lower Viral Loads in Patient Upper Airways. medRxiv 2020. [Google Scholar] [CrossRef]
- Toyoshima, Y.; Nemoto, K.; Matsumoto, S.; Nakamura, Y.; Kiyotani, K. SARS-CoV-2 genomic variations associated with mortality rate of COVID-19. J. Hum. Genet. 2020, 65, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.L.; Sparks, J.S.; Eckerle, L.D.; Sims, A.C.; Denison, M.R. SARS coronavirus replicase proteins in pathogenesis. Virus Res. 2008, 133, 88–100. [Google Scholar] [CrossRef] [PubMed]
- Angeletti, S.; Benvenuto, D.; Bianchi, M.; Giovanetti, M.; Pascarella, S.; Ciccozzi, M. COVID-2019: The role of the nsp2 and nsp3 in its pathogenesis. J. Med. Virol. 2020, 92, 584–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.Y.; Liao, C.H.; Wang, Q.; Tan, Y.J.; Luo, R.; Qiu, Y.; Ge, X.Y. The ORF6, ORF8 and nucleocapsid proteins of SARS-CoV-2 inhibit type I interferon signaling pathway. Virus Res. 2020, 286, 198074. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| SNP | Position | Reference | Variant | X2 | p Value * | OR | OR 95%CI | Protein | AA Change $ |
|---|---|---|---|---|---|---|---|---|---|
| C/T | 1005 | 88.10% | 11.90% | 0.257 | 0.6125 | 1.15 | 0.66–1.99 | nsp2 | T265I |
| C/T | 2362 | 97.60% | 2.40% | 2.602 | 0.1068 | 3.69 | 0.51–26.81 | nsp2 | No change |
| C/T | 2782 | 96.10% | 3.90% | 1.852 | 0.173 | 0.59 | 0.29–1.2 | nsp3 | No change |
| C/T | 2983 | 67.70% | 32.30% | 20.454 | <0.0001 * | 0.38 | 0.24–0.61 | nsp3 | No change |
| C/A | 6258 | 95.80% | 4.20% | 0.177 | 0.674 | 0.82 | 0.33–2.06 | nsp3 | T2016K |
| C/T | 8728 | 90.90% | 9.10% | 9.768 | 0.0018 * | 3.67 | 1.35–10.1 | nsp4 | No change |
| G/T | 11,029 | 87.60% | 12.40% | 11.947 | 0.0005 * | 3.36 | 1.47–7.69 | nsp6 | L3606F |
| C/T | 13,676 | 95.90% | 4.10% | 0.001 | 0.981 | 0.99 | 0.42–2.3 | nsp12 (RdRp) | A4489V |
| T/C | 14,353 | 67.40% | 32.60% | 40.812 | <0.0001 * | 0.22 | 0.13–0.39 | nsp12 (RdRp) | P4714L |
| C/T | 14,751 | 96.80% | 3.20% | 2.075 | 0.149 | 2.45 | 0.6–10.1 | nsp12 (RdRp) | No change |
| C/T | 15,270 | 94.10% | 5.90% | 3.381 | 0.066 | 0.56 | 0.310.99 | nsp12 (RdRp) | No change |
| C/T | 18,823 | 87.30% | 12.70% | 1.377 | 0.241 | 0.75 | 0.47–1.19 | nsp14 | No change |
| A/G | 20,214 | 93.10% | 6.90% | 0.112 | 0.738 | 1.12 | 0.56–2.24 | nsp15 | No change |
| C/T | 22,390 | 94.80% | 5.20% | 4.201 | 0.04 | 0.51 | 0.28–0.93 | S | No change |
| A/G | 23,349 | 31.90% | 68.10% | 38.338 | <0.0001 * | 4.23 | 2.46–7.27 | S | G614D |
| C/T | 23,875 | 96.00% | 4.00% | 0.109 | 0.741 | 1.16 | 0.47–2.91 | S | No change |
| G/T | 25,508 | 72.30% | 27.70% | 0.156 | 0.693 | 0.93 | 0.64–1.35 | Orf3a | Q57H |
| G/T | 26,090 | 95.90% | 4.10% | 6.572 | 0.0104 | 6.37 | 0.88–45.96 | Orf3a | G251V |
| C/T | 26,681 | 91.00% | 9.00% | 1.487 | 0.222 | 0.71 | 0.42–1.2 | M | No change |
| T/C | 28,090 | 90.20% | 9.80% | 11.194 | 0.0008 * | 0.25 | 0.09–0.69 | Orf8 | L84S |
| C/T | 28,257 | 95.70% | 4.30% | 0.199 | 0.656 | 1.22 | 0.49–3.05 | N | P13L |
| C/T | 28,800 | 94.70% | 5.30% | 2.587 | 0.108 | 0.58 | 0.31–1.08 | N | S194L |
| G/A | 28,823 | 97.70% | 2.30% | 6.812 | 0.0091 | 0 | - | N | S202N |
| G/A # | 28,827 | 82.00% | 18.00% | 31.579 | <0.0001 * | 2.93 | 2.05–4.19 | N | R203K G204R |
| G/A # | 28,828 | 82.00% | 18.00% | 32.172 | <0.0001 * | 2.96 | 2.08–4.24 | N | R203K G204R |
| G/C # | 28,829 | 82.00% | 18.00% | 31.444 | <0.0001 * | 2.92 | 2.05–4.18 | N | R203K G204R |
| G/A/T | 29,688 | 95.60% | 2.50% | 12.896 | 0.0016 * | - | - | 3’UTR | - |
| Term | Estimate | Std Error | X2 | p Value | Lower CL | Upper CL |
|---|---|---|---|---|---|---|
| Intercept | −5.103 | 1.143 | 37.51 | <0.0001 * | −7.533 | −3.310 |
| Africa | −1.069 | 0.675 | 5.37 | 0.021 * | −2.836 | 0.030 |
| Asia | 0.198 | 0.229 | 3.90 | 0.048 * | −0.227 | 0.717 |
| Europe | −0.730 | 0.273 | 6.87 | 0.009 * | −1.264 | −0.148 |
| North America | 0.239 | 0.282 | 3.31 | 0.069 | −0.305 | 0.834 |
| Jan | −1.929 | 1.265 | 2.08 | 0.149 | −6.105 | −0.096 |
| Feb | −1.360 | 0.773 | 2.08 | 0.149 | −3.292 | 0.010 |
| Mar | −0.100 | 0.385 | 1.80 | 0.18 | −0.766 | 0.818 |
| Apr | 0.810 | 0.379 | 8.76 | 0.003 * | 0.161 | 1.721 |
| May | 0.317 | 0.454 | 2.16 | 0.142 | −0.541 | 1.320 |
| Jun | 1.161 | 0.439 | 10.10 | 0.002 * | 0.362 | 2.147 |
| Female sex | −0.216 | 0.100 | 9.26 | 0.002 * | −0.419 | −0.019 |
| Age | 0.052 | 0.006 | 108.63 | <0.0001 * | 0.041 | 0.064 |
| C2983 | 1.833 | 0.753 | 11.03 | 0.001 * | 0.690 | 3.585 |
| T2983 | −1.160 | 0.695 | 0.15 | 0.697 | −2.147 | 0.530 |
| C14353 | −2.349 | 0.690 | 0.36 | 0.549 | −3.514 | −0.583 |
| T14353 | 0.767 | 0.587 | 4.77 | 0.029 * | −0.107 | 2.434 |
| AAC28827 | 0.001 | 0.578 | 1.07 | 0.301 | −0.820 | 1.648 |
| GGG28827 | −0.824 | 0.573 | 0.41 | 0.523 | −1.625 | 0.819 |
| SNP | Position | Reference | Variant | X2 | p Value * | Protein |
|---|---|---|---|---|---|---|
| C/T | 2983 | 75.73% | 24.27% | 20.318 | <0.0001 * | nsp3 |
| C/T | 8728 | 89.59% | 10.41% | 8.250 | 0.0041 * | nsp4 |
| G/T/A | 11,029 | 90.07% | 9.93% | 7.633 | 0.022 | nsp6 |
| T/C | 14,353 | 73.71% | 26.29% | 10.845 | 0.0010 * | nsp12 (RdRp) |
| A/G | 23,349 | 23.91% | 76.09% | 20.529 | <0.0001 * | S |
| T/C | 28,090 | 89.53% | 10.47% | 8.303 | 0.0040 * | Orf8 |
| G/A # | 28,827 | 60.96% | 39.04% | 9.395 | 0.0022 * | N |
| G/A # | 28,828 | 60.99% | 39.01% | 9.374 | 0.0022 * | N |
| G/C/A # | 28,829 | 60.44% | 38.94% | 31.444 | 0.0094 | N |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dumonteil, E.; Fusco, D.; Drouin, A.; Herrera, C. Genomic Signatures of SARS-CoV-2 Associated with Patient Mortality. Viruses 2021, 13, 227. https://doi.org/10.3390/v13020227
Dumonteil E, Fusco D, Drouin A, Herrera C. Genomic Signatures of SARS-CoV-2 Associated with Patient Mortality. Viruses. 2021; 13(2):227. https://doi.org/10.3390/v13020227
Chicago/Turabian StyleDumonteil, Eric, Dahlene Fusco, Arnaud Drouin, and Claudia Herrera. 2021. "Genomic Signatures of SARS-CoV-2 Associated with Patient Mortality" Viruses 13, no. 2: 227. https://doi.org/10.3390/v13020227
APA StyleDumonteil, E., Fusco, D., Drouin, A., & Herrera, C. (2021). Genomic Signatures of SARS-CoV-2 Associated with Patient Mortality. Viruses, 13(2), 227. https://doi.org/10.3390/v13020227