
An Efficient, Counter-Selection-Based Method for Prophage Curing in Pseudomonas aeruginosa Strains

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Plasmids and Growth Media
2.2. DNA Manipulation and Plasmid Construction
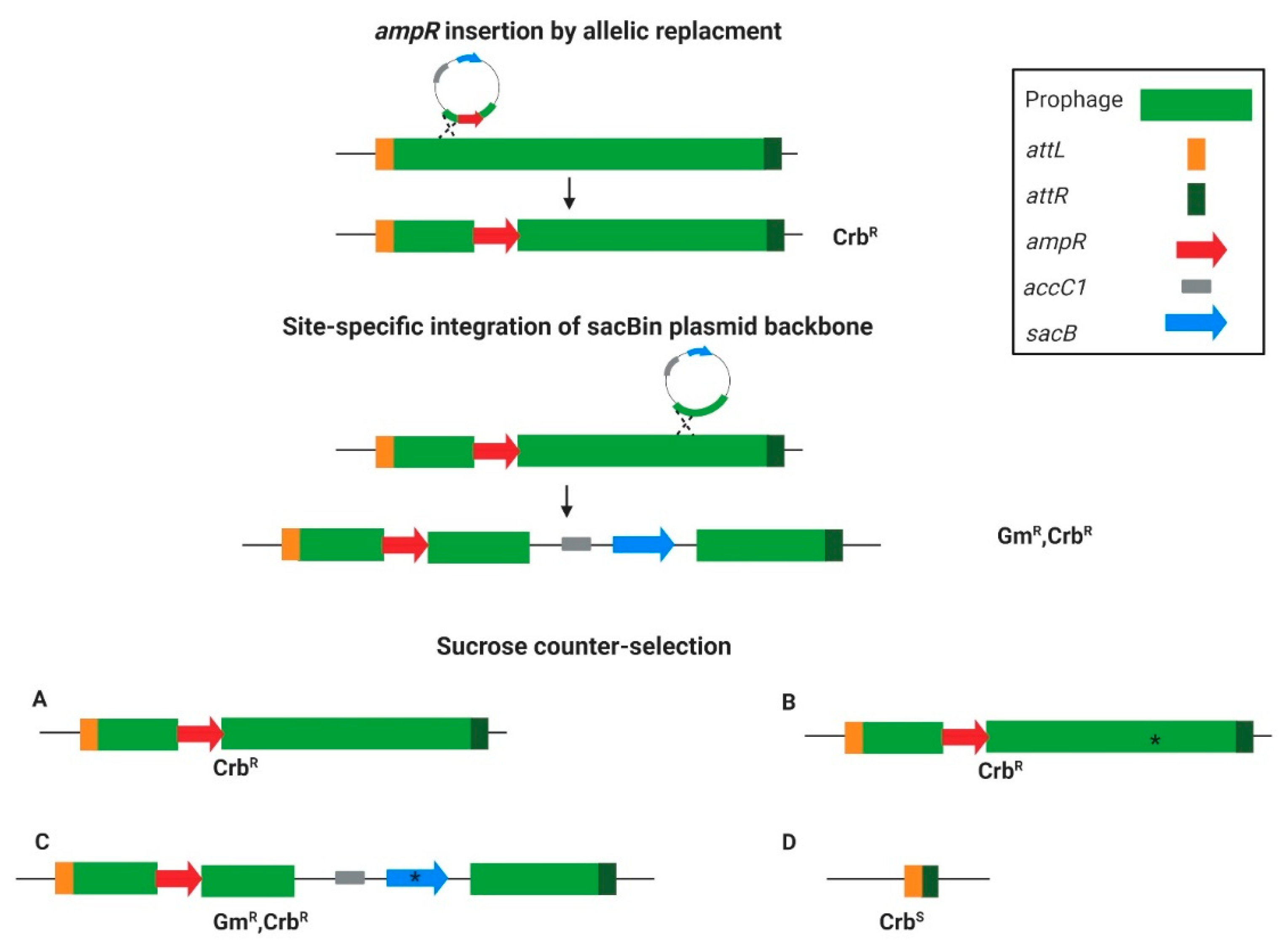
2.3. Insertion of ampR into Phage Region
2.4. Prophage Curing and Selection for Phage-Cured Mutants
2.5. Plasmid Curing
2.6. Phage Extraction
2.7. Plaque Assay
2.8. Whole-Genome Sequencing
2.9. Genome Assembly and Sequence Analysis
3. Results
3.1. The Targeted Curing Principle
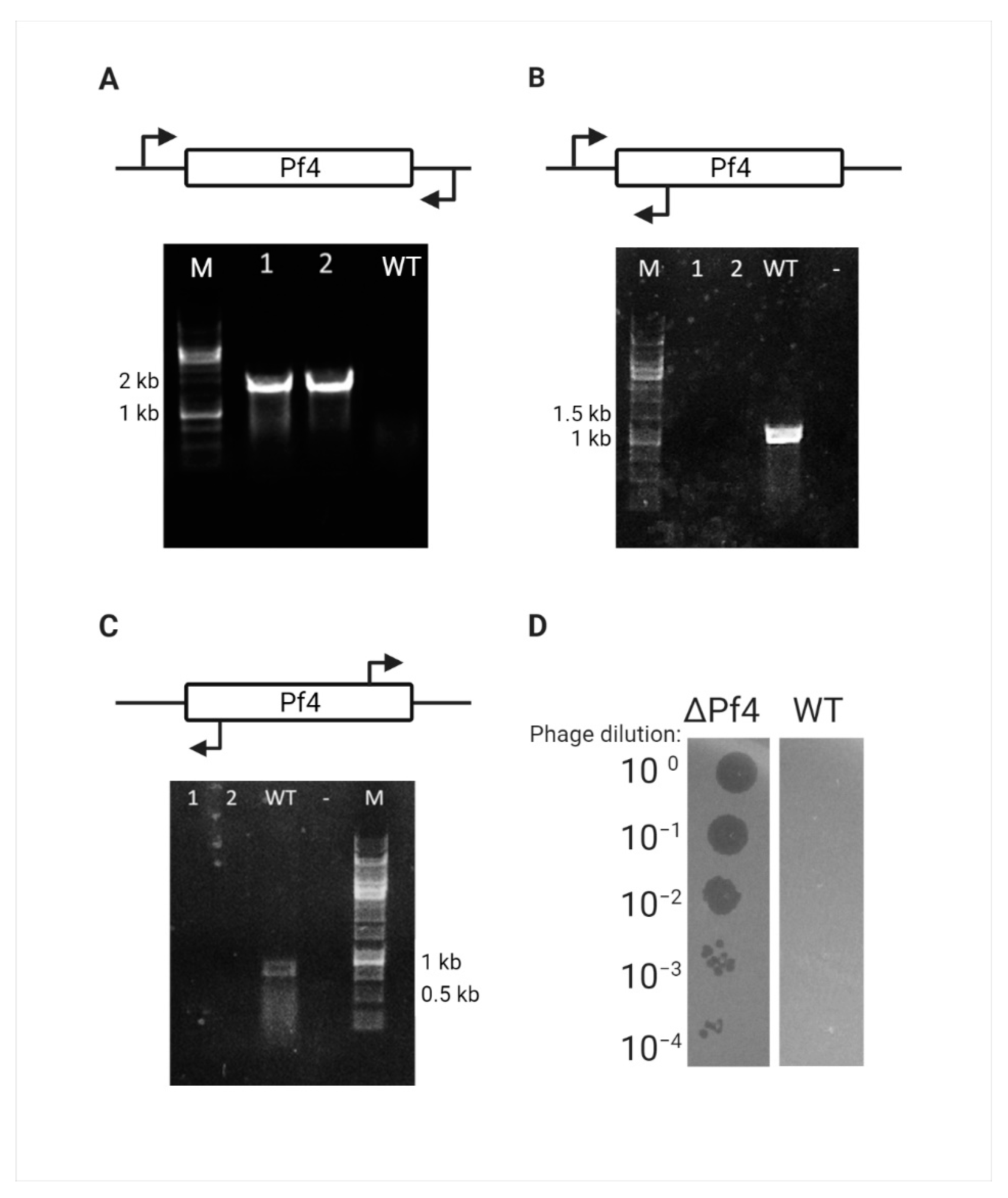
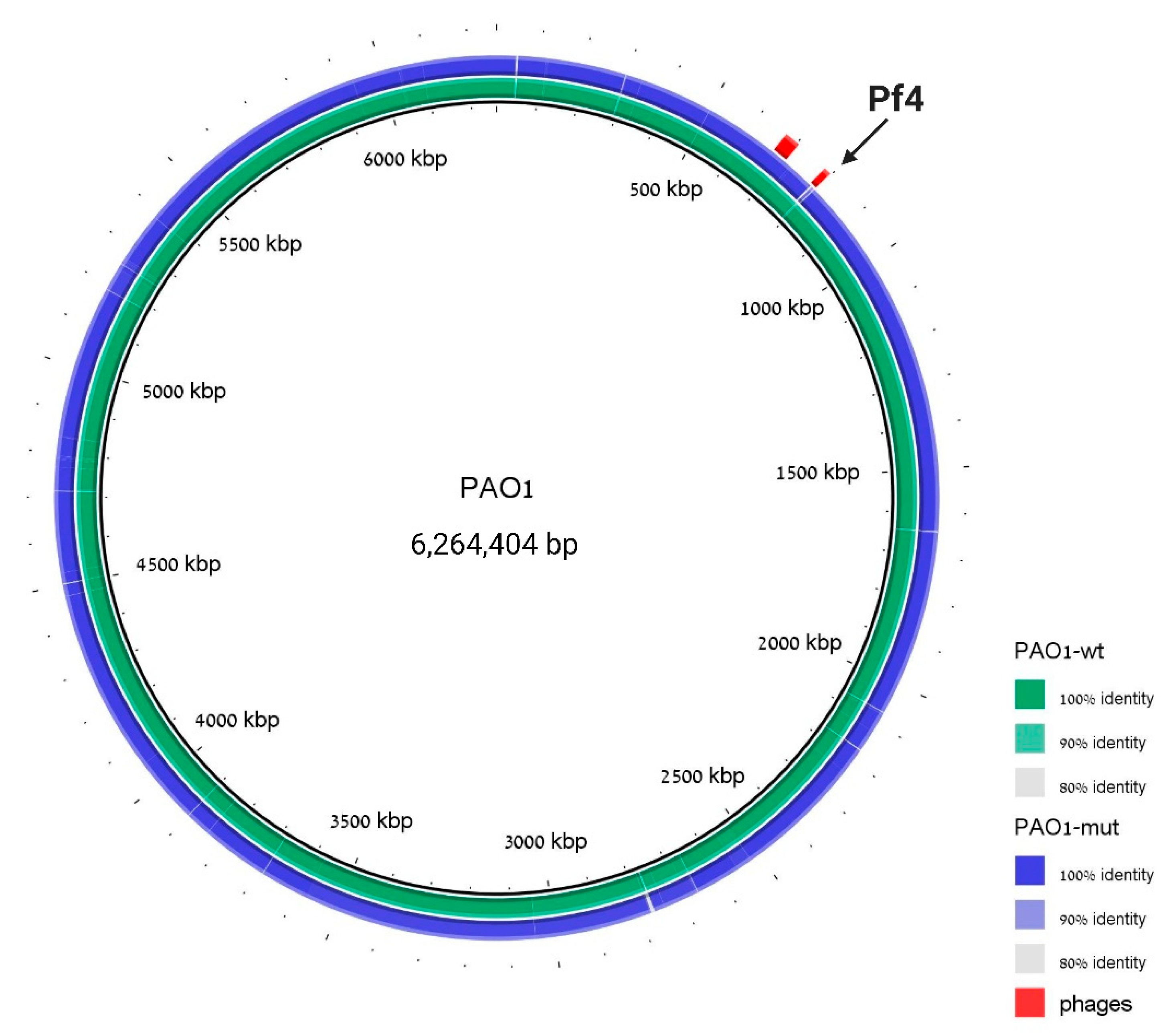
3.2. Pf4 Phage of PAO1 Curing
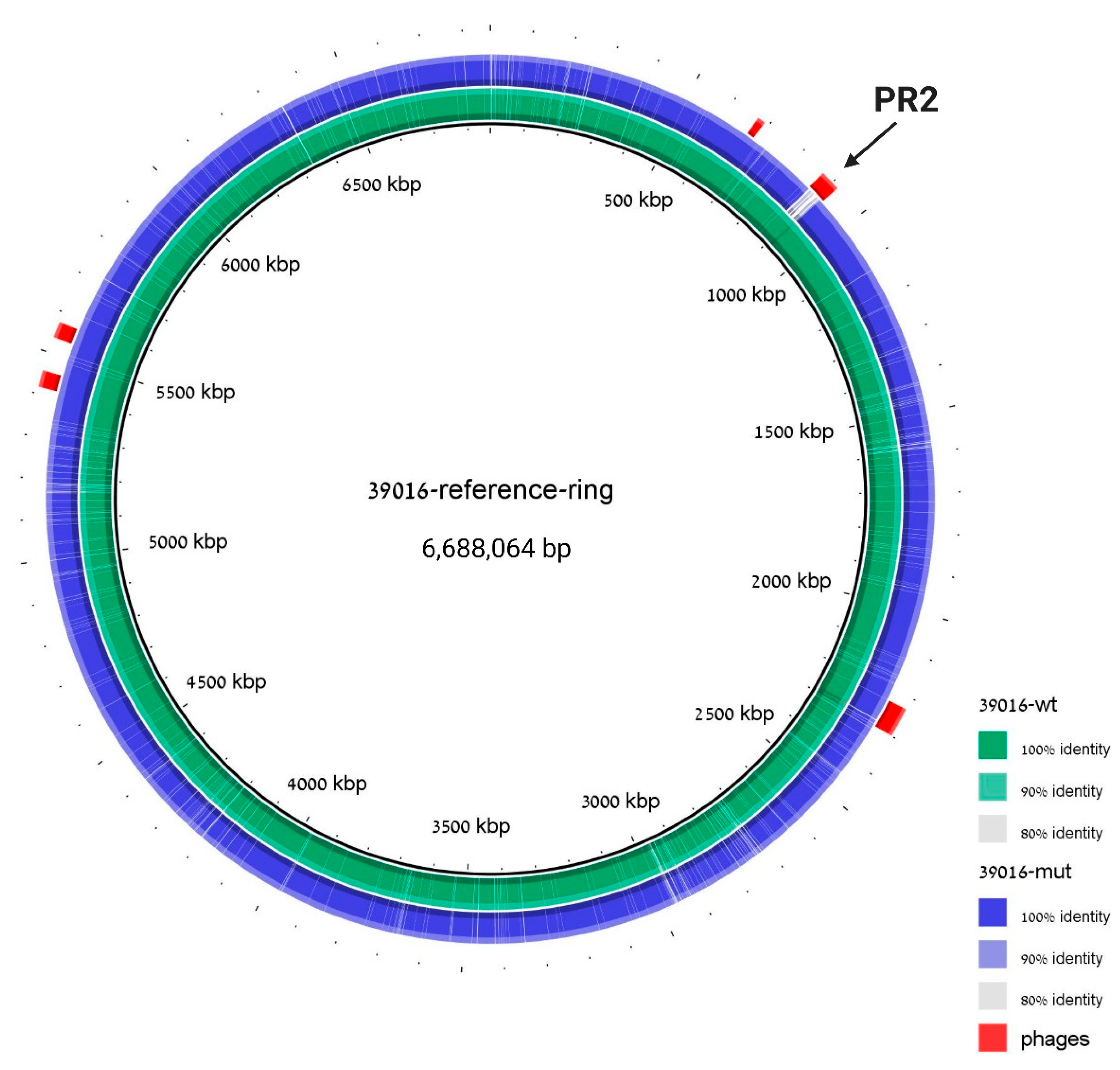
3.3. PR2 Phage of 39016 Curing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pirnay, J.P.; Bilocq, F.; Pot, B.; Cornelis, P.; Zizi, M.; Van Eldere, J.; Deschaght, P.; Vaneechoutte, M.; Jennes, S.; Pitt, T.; et al. Pseudomonas aeruginosa Population Structure Revisited. PLoS ONE 2009, 4, e7740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathee, K.; Lory, S.; Narasimhan, G.; Valdes, C.; Qiu, X.; Matewish, J.M.; Koehrsen, M.; Rokas, A.; Yandava, C.N.; Engels, R.; et al. Dynamics of Pseudomonas aeruginosa Genome Evolution. Proc. Natl. Acad. Sci. USA 2008, 105, 3100–3105. [Google Scholar] [CrossRef] [Green Version]
- James, C.E.; Fothergill, J.L.; Kalwij, H.; Hall, A.J.; Cottell, J.; Brockhurst, M.A.; Winstanley, C. Differential Infection Properties of Three Inducible Prophages from an Epidemic Strain of Pseudomonas aeruginosa. BMC Microbiol. 2012, 12, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aloush, V.; Navon-Venezia, S.; Seigman-Igra, Y.; Cabili, S.; Carmeli, Y. Multidrug-Resistant Pseudomonas aeruginosa: Risk Factors and Clinical Impact. Antimicrob. Agents Chemother. 2006, 50, 43–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gellatly, S.L.; Hancock, R.E.W. Pseudomonas aeruginosa: New Insights into Pathogenesis and Host Defenses. Pathog. Dis. 2013, 67, 159–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiehlmann, L.; Wagner, G.; Cramer, N.; Siebert, B.; Gudowius, P.; Morales, G.; Kohler, T.; van Delden, C.; Weinel, C.; Slickers, P.; et al. Population Structure of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2007, 104, 8101–8106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payet, J.P.; Suttle, C.A. To Kill or Not to Kill: The Balance between Lytic and Lysogenic Viral Infection Is Driven by Trophic Status. Limnol. Oceanogr. 2013, 58, 465–474. [Google Scholar] [CrossRef]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in Nature: Mechanisms, Impact and Ecology of Temperate Phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canchaya, C.; Proux, C.; Fournous, G.; Bruttin, A.; Brüssow, H. Prophage Genomics. Microbiol. Mol. Biol. Rev. 2003, 67, 238–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.C.; Levy, S.B.; Jackson, R.W. Pseudomonas Genomes: Diverse and Adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knezevic, P.; Voet, M.; Lavigne, R. Prevalence of Pf1-like (pro)Phage Genetic Elements among Pseudomonas aeruginosa Isolates. Virology 2015, 483, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rice, S.A.; Tan, C.H.; Mikkelsen, P.J.; Kung, V.; Woo, J.; Tay, M.; Hauser, A.; McDougald, D.; Webb, J.S.; Kjelleberg, S. The Biofilm Life Cycle and Virulence of Pseudomonas aeruginosa Are Dependent on a Filamentous Prophage. ISME J. 2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzio, J.; Kropinski, A.M. O-Antigen Conversion in Pseudomonas aeruginosa PAO1 by Bacteriophage D3. J. Bacteriol. 1983. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Baba, T.; Matsumoto, H.; Terawaki, Y. Phage-conversion of Cytotoxin Production in Pseudomonas aeruginosa. Mol. Microbiol. 1990. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.S.; Lau, M.; Kjelleberg, S. Bacteriophage and Phenotypic Variation in Pseudomonas aeruginosa Biofilm Development. J. Bacteriol. 2004, 186, 8066–8073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Secor, P.R.; Sweere, J.M.; Michaels, L.A.; Malkovskiy, A.V.; Lazzareschi, D.; Katznelson, E.; Rajadas, J.; Birnbaum, M.E.; Arrigoni, A.; Braun, K.R.; et al. Filamentous Bacteriophage Promote Biofilm Assembly and Function. Cell Host Microbe 2015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zander, I. Identifying and Examining the Effect of Bacteriophage Derived Genes on Pseudomonas aeruginosa Virulence and Physiology. unpublished.
- Novick, R. Properties of a Cryptic High-Frequency Transducing Phage in Staphylococcus aureus. Virology 1967. [Google Scholar] [CrossRef]
- Wiederholt, K.M.; Steele, J.L. Prophage Curing and Partial Characterization of Temperate Bacteriophages from Thermolytic Strains of Lactococcus lactis ssp. Cremoris. J. Dairy Sci. 1993, 76, 921–930. [Google Scholar] [CrossRef]
- Gay, P.; Le Coq, D.; Steinmetz, M.; Berkelman, T.; Kado, C.I. Positive Selection Procedure for Entrapment of Insertion Sequence Elements in Gram-Negative Bacteria. J. Bacteriol. 1985. [Google Scholar] [CrossRef] [Green Version]
- Hmelo, L.R.; Hmelo, B.R.; Almblad, H.; Love, M.E.; Randall, T.E.; Tseng, B.S.; Lin, C.; Irie, Y.; Storek, K.M.; Yang, J.J.; et al. Precision-Engineering the Pseudomonas aeruginosa Genome with Two-Step Allelic Exchange. Nat. Rev. Drug Discov. 2016, 5, 1–8. [Google Scholar] [CrossRef]
- Wozniak, D.J.; Ohman, D.E. Pseudomonas aeruginosa AlgB, a Two-Component Response Regulator of the NtrC Family, Is Required for AlgD Transcription. J. Bacteriol. 1991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zander, I.; Shmidov, E.; Roth, S.; Ben-David, Y.; Shoval, I.; Shoshani, S.; Danielli, A.; Banin, E. Characterization of PfiT/PfiA Toxin-antitoxin System of Pseudomonas aeruginosa That Affects Cell Elongation and Prophage Induction. Environ. Microbiol. 2020. [Google Scholar] [CrossRef]
- Simon, R.; Priefer, U.; Puhler, A. A Broad Host Range Mobilization System for in Vivo Genetic Engineering: Transposon Mutagenesis in Gram Negative Bacteria. Nat. Biotechnol. 1983, 1, 784–791. [Google Scholar] [CrossRef]
- Schweizer, H.P. Escherichia-Pseudomonas Shuttle Vectors Derived from PUC18/19. Gene 1991, 97, 109–112. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013. [Google Scholar] [CrossRef] [PubMed]
- Rissman, A.I.; Mau, B.; Biehl, B.S.; Darling, A.E.; Glasner, J.D.; Perna, N.T. Reordering Contigs of Draft Genomes Using the Mauve Aligner. Bioinformatics 2009. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T. Progressivemauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A Better, Faster Version of the PHAST Phage Search Tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alikhan, N.F.; Petty, N.K.; Ben Zakour, N.L.; Beatson, S.A. BLAST Ring Image Generator (BRIG): Simple Prokaryote Genome Comparisons. BMC Genomics 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai-Prochnow, A.; Hui, J.G.K.; Kjelleberg, S.; Rakonjac, J.; McDougald, D.; Rice, S.A. “Big Things in Small Packages: The Genetics of Filamentous Phage and Effects on Fitness of Their Host”. FEMS Microbiol. Rev. 2015, 39, 465–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Euler, C.W.; Juncosa, B.; Ryan, P.A.; Deutsch, D.R.; McShan, W.M.; Fischetti, V.A. Targeted Curing of All Lysogenic Bacteriophage from Streptococcus pyogenes Using a Novel Counter-Selection Technique. PLoS ONE 2016, 11, e0146408. [Google Scholar] [CrossRef] [PubMed]
- Ward, H.; Perron, G.G.; MacLean, R.C. The Cost of Multiple Drug Resistance in Pseudomonas aeruginosa. J. Evol. Biol. 2009, 22, 997–1003. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.P.; Ozer, E.A.; Minasov, G.; Shuvalova, L.; Kiryukhina, O.; Satchell, K.J.F.; Hauser, A.R. A Comparative Genomics Approach Identifies Contact-Dependent Growth Inhibition as a Virulence Determinant. Proc. Natl. Acad. Sci. USA 2020. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Rifo, A.; Veksler-Lublinsky, I.; Cheng, Z.; Ausubel, F.M.; Ambros, V. The Pseudomonas aeruginosa Accessory Genome Elements Influence Virulence towards Caenorhabditis Elegans. Genome Biol. 2019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlsen, C.; Hjerde, E.; Klemetsen, T.; Willassen, N.P. Pan Genome and CRISPR Analyses of the Bacterial Fish Pathogen Moritella Viscosa. BMC Genom. 2017. [Google Scholar] [CrossRef]
- Pasechnek, A.; Rabinovich, L.; Stadnyuk, O.; Azulay, G.; Mioduser, J.; Argov, T.; Borovok, I.; Sigal, N.; Herskovits, A.A. Active Lysogeny in Listeria monocytogenes Is a Bacteria-Phage Adaptive Response in the Mammalian Environment. Cell Rep. 2020. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain or plasmid | Description | Source |
|---|---|---|
| P. aeruginosa strains | ||
| PA01 | WT | [22] |
| PAO1 ΔPf4 | PAO1, ΔPf4 | This study |
| PAO1 ΔpfiT | PAO1, ΔPAO729::Crbr | [23] |
| 39016 | LMG 27,647 P. aeruginosa (Schoeter 1872) migula 1900 AL | BCCM-biological origin: keratitis patients |
| 39016/PR2_ampR | 39016, ΔPA39016_000100015::Crbr | This study |
| 30916 ΔPR2/Gmr | 39016, ΔPR2::Gmr | This study |
| 30916 ΔPR2 | 39016, ΔPR2 | This study |
| E. coli strains | ||
| DH5α | F- Φ80lacZΔM15 Δ(lacZYA-argF) U169 recA1 endA1 hsdR17 (rK-, mK+) phoA supE44 λ- thi-1 gyrA96 relA1. | Bio-Lab |
| S17 | E. coli S17 thi, pro, hsdR, recA::RP4 -2-Tc::Mu aphA::Tn7, λ-pir, Smr, Tpr | [24] |
| Plasmids | ||
| pUCP18-Ap | Crbr (for P. aeruginosa), Ampr (for E. coli), overexpression plasmid, lacZ promoter | [25] |
| pDONRPEX18Gm | Gmr and Cmr, pEX18Gm containing a HindIII flanked, attP cloning site from pDONR201 | [21] |
| pDONER/AmpRin_PR2 | Cmr and Gmr, PA39016_000100015 upstream and downstream fragments with AmpR cassette inserted into pDONRPEX18Gm by Gateway recombination | This study |
| pDONER/PR2_SacB | Cmr and Gmr, PA39016_000100043 and PA39016_000100035 fragments inserted into pDONRPEX18Gm by Gateway recombination | This study |
| pDONER/Pf4_SacB | Cmr and Gmr, PA0725 upstream and downstream fragments inserted into pDONRPEX18Gm by Gateway recombination | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shmidov, E.; Zander, I.; Lebenthal-Loinger, I.; Karako-Lampert, S.; Shoshani, S.; Banin, E. An Efficient, Counter-Selection-Based Method for Prophage Curing in Pseudomonas aeruginosa Strains. Viruses 2021, 13, 336. https://doi.org/10.3390/v13020336
Shmidov E, Zander I, Lebenthal-Loinger I, Karako-Lampert S, Shoshani S, Banin E. An Efficient, Counter-Selection-Based Method for Prophage Curing in Pseudomonas aeruginosa Strains. Viruses. 2021; 13(2):336. https://doi.org/10.3390/v13020336
Chicago/Turabian StyleShmidov, Esther, Itzhak Zander, Ilana Lebenthal-Loinger, Sarit Karako-Lampert, Sivan Shoshani, and Ehud Banin. 2021. "An Efficient, Counter-Selection-Based Method for Prophage Curing in Pseudomonas aeruginosa Strains" Viruses 13, no. 2: 336. https://doi.org/10.3390/v13020336
APA StyleShmidov, E., Zander, I., Lebenthal-Loinger, I., Karako-Lampert, S., Shoshani, S., & Banin, E. (2021). An Efficient, Counter-Selection-Based Method for Prophage Curing in Pseudomonas aeruginosa Strains. Viruses, 13(2), 336. https://doi.org/10.3390/v13020336