
Virus Prospecting in Crickets—Discovery and Strain Divergence of a Novel Iflavirus in Wild and Cultivated Acheta domesticus

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design
2.2. Sample Origin and Processing
2.3. Target-Free Screening by Sequencing
2.4. Full Genomes of Wild and Reared AdIV Strains by Sanger Sequencing
2.5. Phylogenetic Analyses
2.6. Targeted Screening for AdIV by RT-qPCR
3. Results
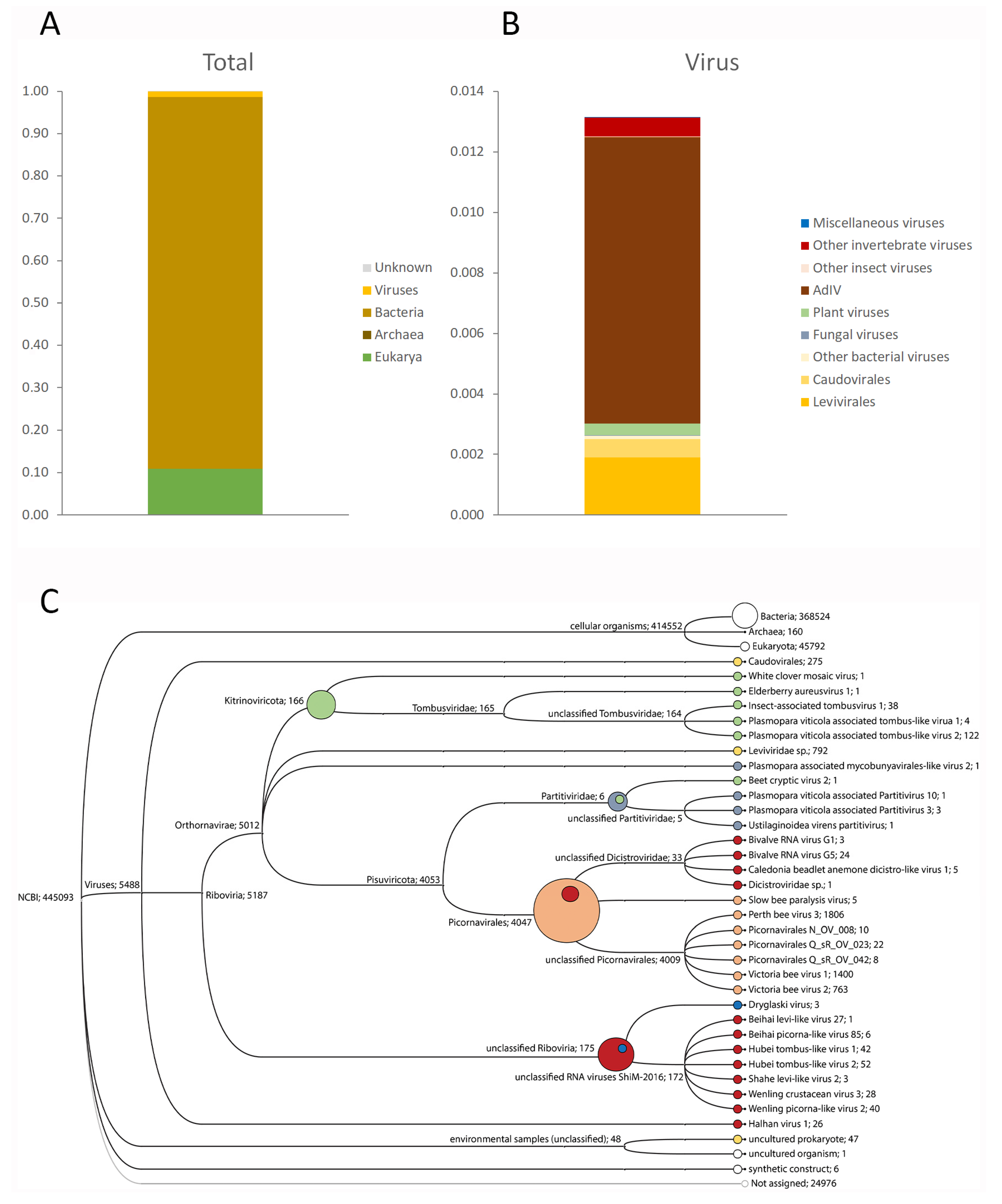
3.1. Target-Free Screening of Acheta Domesticus Frass DNA and RNA
3.2. Genetic Analysis of AdIV Strains from Wild and Commercially Reared Crickets
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.; Chen, X.; Li, C.; Qin, X.; Li, J.; Cao, P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.M.K.; Anderson, D.L.; Durr, P.A. Metagenomic analysis of Varroa-free Australian honey bees (Apis mellifera) shows a diverse Picornavirales virome. J. Gen. Virol. 2018, 99, 818–826. [Google Scholar] [CrossRef] [PubMed]
- Fredricks, D.N.; Relman, D.A. Sequence-based identification of microbial pathogens: A reconsideration of Koch’s postulates. Clin. Microbiol. Rev. 1996, 9, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Casadevall, A.; Pirofski, L.A. What is a host? Incorporating the microbiota into the damage-response framework. Infect. Immun. 2015, 83, 2–7. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.J. Move over, bacteria! Viruses make their mark as mutualistic microbial symbionts. J. Virol. 2015, 89, 6532–6535. [Google Scholar] [CrossRef] [Green Version]
- Beaurepaire, A.; Piot, N.; Doublet, V.; Antuñez, K.; Campbell, E.; Chantawannakul, P.; Chejanovsky, N.; Gajda, A.; Heerman, M.; Panzier, D.; et al. Diversity and global distribution of viruses of the western honey bee, Apis mellifera. Insects 2020, 11, 239. [Google Scholar] [CrossRef] [PubMed]
- Yañez, O.; Piot, N.; Dalmon, A.; de Miranda, J.R.; Chantawannakul, P.; Panziera, D.; Amiri, E.; Smagghe, G.; Schroeder, D.C.; Chejanovsky, N. Bee viruses: Routes of infection in Hymenoptera. Front. Microbiol. 2020, 11, e943. [Google Scholar] [CrossRef]
- Martin, S.J.; Brettell, L.E. Deformed wing virus in honeybees and other insects. Annu. Rev. Virol. 2019, 6, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Traynor, K.S.; Mondet, F.; de Miranda, J.R.; Techer, M.; Kowallik, V.; Oddie, M.A.Y.; Chantawannakul, P.; McAfee, A. Varroa destructor: A complex parasite, crippling honeybees worldwide. Trends Parasitol. 2020, 36, 592–606. [Google Scholar] [CrossRef]
- Dolan, P.T.; Whitfield, Z.J.; Andino, R. Mechanisms and concepts in RNA virus population dynamics and evolution. Annu. Rev. Virol. 2018, 5, 69–92. [Google Scholar] [CrossRef]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [Green Version]
- Yañez, O.; Chávez-Galarza, J.; Tellgren-Roth, C.; Pinto, M.A.; Neumann, P.; de Miranda, J.R. The honeybee (Apis mellifera) developmental state shapes the genetic composition of the deformed wing virus-A quasispecies during serial transmission. Sci. Rep. 2020, 10, e5956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ongus, J.R.; Peters, D.; Bonmatin, J.M.; Bengsch, E.; Vlak, J.M.; van Oers, M.M. Complete sequence of a picorna-like virus of the genus Iflavirus replicating in the mite Varroa destructor. J. Gen. Virol. 2004, 85, 3747–3755. [Google Scholar] [CrossRef]
- Gisder, S.; Möckel, N.; Eisenhardt, D.; Genersch, E. In vivo evolution of viral virulence: Switching of deformed wing virus between hosts results in virulence changes and sequence shifts. Environ. Microbiol. 2018, 20, 4612–4628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryabov, E.V.; Childers, A.K.; Lopez, D.; Grubbs, K.; Posada-Florez, F.; Weaver, D.; Evans, J.D. Dynamic evolution in the key honey bee pathogen deformed wing virus: Novel insights into virulence and competition using reverse genetics. PLoS Biol. 2019, 17, e3000502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E. ICTV Report Consortium. ICTV Virus Taxonomy Profile: Iflaviridae. J. Gen. Virol. 2017, 98, 527–528. [Google Scholar] [CrossRef]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E. ICTV Report Consortium. ICTV Virus Taxonomy Profile: Dicistroviridae. J. Gen. Virol. 2017, 98, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Genersch, E. Honey bee pathology: Current threats to honey bees and beekeeping. Appl. Microbiol. Biotechnol. 2010, 87, 87–97. [Google Scholar] [CrossRef]
- Kang, G.P.; Guo, X.J. Overview of silkworm pathology in China. Afr. J. Biotechnol. 2011, 10, 18046–18056. [Google Scholar]
- Dobermann, D. Insects as food and feed: Can research and business work together? J. Insect. Food Feed 2017, 3, 155–160. [Google Scholar] [CrossRef]
- Berggren, Å.; Jansson, A.; Low, M. Using current systems to inform rearing facility design in the insect-as-food industry. J. Insect. Food Feed 2018, 4, 167–170. [Google Scholar] [CrossRef]
- Maciel-Vergara, G.; Ros, V.I.D. Viruses of insects reared for food and feed. J. Invertebr. Pathol. 2017, 147, 60–75. [Google Scholar] [CrossRef]
- Eilenberg, J.; Vlak, J.M.; Nielsen-LeRoux, C.; Cappellozza, S.; Jensen, A.B. Diseases in insects produced for food and feed. J. Insect. Food Feed 2015, 1, 87–102. [Google Scholar] [CrossRef] [Green Version]
- van Huis, A. Edible crickets, but which species? J. Insect. Food Feed 2020, 6, 91–94. [Google Scholar] [CrossRef]
- van Huis, A. Insects as food and feed, a new emerging agricultural sector: A review. J. Insect. Food Feed 2020, 6, 27–44. [Google Scholar] [CrossRef] [Green Version]
- Pippinato, L.; Gasco, L.; Di Vita, G.; Mancuso, T. Current scenario in the European edible-insect industry: A preliminary study. J. Insect. Food Feed 2020, 6, 371–381. [Google Scholar] [CrossRef]
- van Huis, A.; Tomberlin, J.K. Future prospects. In Insects as Food and Feed: From Production to Consumption; van Huis, A., Tomberlin, J.K., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2017; pp. 430–445. [Google Scholar]
- Semberg, E.; de Miranda, J.R.; Low, M.; Jansson, A.; Forsgren, E.; Berggren, Å. Diagnostic protocols for the detection of Acheta domesticus densovirus (AdDV) in cricket frass. J. Virol. Meth. 2019, 264, 61–64. [Google Scholar] [CrossRef]
- Picard Toolkit. Broad Institute. GitHub Repository. 2019. Available online: http://broadinstitute.github.io/picard/ (accessed on 3 June 2020).
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Babraham Bioinformatics Group. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 3 June 2020).
- Gordon, A.; Hannon, G.J. Fastx-Toolkit: FASTQ/A Short-Reads Pre-Processing Tools. 2010. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 3 June 2020).
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition—Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [Green Version]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, e385. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comp. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [CrossRef]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum information for publication of quantitative Real-Time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, J.D.; Schwarz, R.S.; Chen, Y.-P.; Budge, G.; Cornman, R.S.; De La Rua, P.; de Miranda, J.R.; Foret, S.; Foster, L.; Gauthier, L.; et al. Standard methodologies for molecular research in Apis mellifera. J. Apic. Res. 2013, 52. [Google Scholar] [CrossRef] [Green Version]
- Guo, C.; McDowell, I.C.; Nodzenski, M.; Scholtens, D.M.; Allen, A.S.; Lowe, W.L.; Reddy, T.E. Transversions have larger regulatory effects than transitions. BMC Genom. 2017, 18, e394. [Google Scholar] [CrossRef] [Green Version]
- Koch, R. Uber bakteriologische Forschung. In Proceedings of the Verhandlung des X Internationalen Medichinischen Congresses, Berlin, Germany, 4–9 August 1890; Verlag von August Hirschwald: Berlin, Germany, 1981; Volume 1, p. 35. [Google Scholar]
- de Miranda, J.R.; Hedman, H.; Onorati, P.; Stephan, J.; Karlberg, O.; Bylund, H.; Terenius, O. Characterization of a novel RNA virus discovered in the autumnal moth Epirrita autumnata in Sweden. Viruses 2017, 9, 214. [Google Scholar] [CrossRef]
- de Miranda, J.R.; Cornman, R.S.; Evans, J.D.; Semberg, E.; Haddad, N.; Neumann, P.; Gauthier, L. Genome characterization, prevalence and distribution of a Macula-like virus from Apis mellifera and Varroa destructor. Viruses 2015, 7, 3586–3602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dheilly, N.M.; Maure, F.; Ravallec, M.; Galinier, R.; Doyon, J.; Duval, D.; Leger, L.; Volkoff, A.N.; Missé, D.; Nidelet, S.; et al. Who is the puppet master? Replication of a parasitic wasp-associated virus correlates with host behaviour manipulation. Proc. Biol. Sci. 2015, 282, e20142773. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.J. Mechanisms of plant virus evolution. Annu. Rev. Phytopathol. 1997, 35, 191–209. [Google Scholar] [CrossRef] [PubMed]
- Lyons, D.M.; Lauring, A.S. Evidence for the selective basis of Transition-to-Transversion substitution bias in two RNA viruses. Mol. Biol. Evol. 2017, 34, 3205–3215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanzi, G.; de Miranda, J.R.; Boniotti, M.B.; Cameron, C.E.; Lavazza, A.; Capucci, L.; Camazine, S.M.; Rossi, C. Molecular and biological characterization of deformed wing virus of honeybees (Apis mellifera L). J. Virol. 2006, 80, 4998–5009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Miranda, J.R.; Dainat, B.; Locke, B.; Cordoni, G.; Berthoud, H.; Gauthier, L.; Neumann, P.; Budge, G.E.; Ball, B.V.; Stoltz, D.B. Genetic characterization of slow bee paralysis virus of the honeybee (Apis mellifera L.). J. Gen. Virol. 2010, 91, 2524–2530. [Google Scholar] [CrossRef] [PubMed]
- Procházková, M.; Škubník, K.; Füzik, T.; Mukhamedova, L.; Přidal, A.; Plevka, P. Virion structures and genome delivery of honeybee viruses. Curr. Opin. Virol. 2020, 45, 17–24. [Google Scholar] [CrossRef]
- de Miranda, J.R.; Genersch, E. Deformed wing virus. J. Invertebr. Pathol. 2010, 103, S48–S61. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, R.C.; Ball, B.V.; Willcocks, M.M.; Carter, M.J. The nucleotide sequence of sacbrood virus of the honey bee: An insect picorna-like virus. J. Gen. Virol. 1999, 80, 1541–1549. [Google Scholar] [CrossRef]
- Guan, H.; Tian, J.; Qin, B.; Wojdyla, J.A.; Wang, B.; Zhao, Z.; Wang, M.; Cui, S. Crystal structure of 2C helicase from enterovirus 71. Sci. Adv. 2017, 3, e1602573. [Google Scholar] [CrossRef] [Green Version]
- Černý, J.; Černá Bolfíková, B.; Valdés, J.J.; Grubhoffer, L.; Růžek, D. Evolution of tertiary structure of viral RNA dependent polymerases. PLoS ONE 2014, 9, e96070. [Google Scholar] [CrossRef] [Green Version]
- Venkataraman, S.; Prasad, B.V.L.S.; Selvarajan, R. RNA dependent RNA polymerases: Insights from structure, function and evolution. Viruses 2018, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandez-Cassi, X.; Söderqvist, K.; Bakeeva, A.; Vaga, M.; Dicksved, J.; Vagsholm, I.; Jansson, A.; Boqvist, S. Microbial communities and food safety aspects of crickets (Acheta domesticus) reared under controlled conditions. J. Insect. Food Feed 2020, 6, 429–440. [Google Scholar] [CrossRef]
- Geng, P.; Li, W.; Lin, L.; de Miranda, J.R.; Emrich, S.; An, L.; Terenius, O. Genetic characterization of a novel Iflavirus associated with vomiting disease in the Chinese oak silkmoth Antheraea pernyi. PLoS ONE 2014, 9, e92107. [Google Scholar] [CrossRef] [Green Version]
- Ryabov, E.V. A novel virus isolated from the aphid Brevicoryne brassicae with similarity to Hymenoptera picorna-like viruses. J. Gen. Virol. 2007, 88, 2590–2595. [Google Scholar] [CrossRef]
- Calla, B.; Hall, B.; Hou, S.; Geib, S.M. A genomic perspective to assessing quality of mass-reared SIT flies used in Mediterranean fruit fly (Ceratitis capitata) eradication in California. BMC Genom. 2014, 15, e98. [Google Scholar] [CrossRef] [Green Version]
- Mordecai, G.J.; Wilfert, L.; Martin, S.J.; Jones, I.M.; Schroeder, D.C. Diversity in a honey bee pathogen: First report of a third master variant of the Deformed Wing Virus quasispecies. ISME J. 2016, 10, 1264–1273. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Zhang, J.; Lu, J.; Yi, F.; Liu, C.; Hu, Y. Sequence analysis and genomic organization of a new insect picorna-like virus, Ectropis obliqua picorna-like virus, isolated from Ectropis obliqua. J. Gen. Virol. 2004, 85, 1145–1151. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, S.; Bonning, B.C. Genome sequence of a novel Iflavirus from the leafhopper Graminella nigrifrons. Genome Announc. 2015, 3, e00323-15. [Google Scholar] [CrossRef] [Green Version]
- Isawa, H.; Asano, S.; Sahara, K.; Iizuka, T.; Bando, H. Analysis of genetic information of an insect picorna-like virus, infectious flacherie virus of silkworm: Evidence for evolutionary relationships among insect, mammalian and plant picorna (-like) viruses. Arch. Virol. 1998, 143, 127–143. [Google Scholar] [CrossRef]
- Nakao, R.; Matsuno, K.; Qiu, Y.; Maruyama, J.; Eguchi, N.; Nao, N.; Kajihara, M.; Yoshii, K.; Sawa, H.; Takada, A.; et al. Putative RNA viral sequences detected in an Ixodes scapularis-derived cell line. Ticks Tick Borne Dis. 2017, 8, 103–111. [Google Scholar] [CrossRef] [Green Version]
- Webster, C.L.; Waldron, F.M.; Robertson, S.; Crowson, D.; Ferrari, G.; Quintana, J.F.; Brouqui, J.M.; Bayne, E.H.; Longdon, B.; Buck, A.H.; et al. The discovery, distribution, and evolution of viruses associated with Drosophila melanogaster. PLoS Biol. 2015, 13, e1002210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, O.P.; Snodgrass, G.L.; Allen, K.C.; Jackson, R.E.; Becnel, J.J.; O’Leary, P.F.; Luttrell, R.G. The complete genome sequence of a single-stranded RNA virus from the tarnished plant bug, Lygus lineolaris (Palisot de Beauvois). J. Invertebr. Pathol. 2012, 109, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Suetsugu, Y.; Kobayashi, T.; Nakashima, N. The genome sequence and transmission of an iflavirus from the brown planthopper, Nilaparvata lugens. Virus Res. 2013, 176, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Murakami, R.; Suetsugu, Y.; Nakashima, N. Complete genome sequences of two iflaviruses from the brown planthopper, Nilaparvata lugens. Arch. Virol. 2014, 159, 585–588. [Google Scholar] [CrossRef]
- Wu, C.Y.; Lo, C.F.; Huang, C.J.; Yu, H.T.; Wang, C.H. The complete genome sequence of Perina nuda picorna-like virus, an insect-infecting RNA virus with a genome organization similar to that of the mammalian picornaviruses. Virology 2002, 294, 312–323. [Google Scholar] [CrossRef] [Green Version]
- Choe, S.E.; Nguyen, T.T.; Hyun, B.H.; Noh, J.H.; Lee, H.S.; Lee, C.H.; Kang, S.W. Genetic and phylogenetic analysis of South Korean sacbrood virus isolates from infected honey bees (Apis cerana). Vet. Microbiol. 2012, 157, 32–40. [Google Scholar] [CrossRef]
- Millán-Leiva, A.; Jakubowska, A.K.; Ferré, J.; Herrero, S. Genome sequence of SeIV-1, a novel virus from the Iflaviridae family infective to Spodoptera exigua. J. Invertebr. Pathol. 2012, 109, 127–133. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| SampleID | Supplier | Date | Species | Stage | Type | AdIV |
|---|---|---|---|---|---|---|
| AD:0se | Wild | 2017-09-26 | Acheta domesticus | adult | frass | +++ |
| AD:0dk | A | 2017-05-03 | Acheta domesticus | adult | frass | + |
| AD:057 | B | 2020-01-22 | Gryllus bimaculatus | adult | insect | - |
| AD:061 | B | 2020-01-22 | Gryllus bimaculatus | adult | insect | - |
| AD:065 | B | 2020-01-22 | Gryllus bimaculatus | adult | frass | - |
| AD:069 | C | 2020-06-28 | Acheta domesticus | nymph | insect | - |
| AD:072 | C | 2020-06-29 | Acheta domesticus | nymph | frass | +++ |
| AD:073 | C | 2020-06-29 | Acheta domesticus | juvenile | frass | ++ |
| AD:074 | C | 2020-06-29 | Acheta domesticus | adult | frass | - |
| AD:075 | D | 2020-09-09 | Acheta domesticus | adult | insect | - |
| AD:076 | D | 2020-09-09 | Acheta domesticus | adult | insect | ++ |
| AD:077 | D | 2020-09-09 | Acheta domesticus | adult | insect | ++ |
| AD:078 | E | 2020-09-25 | Acheta domesticus | nymph | insect | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Miranda, J.R.; Granberg, F.; Onorati, P.; Jansson, A.; Berggren, Å. Virus Prospecting in Crickets—Discovery and Strain Divergence of a Novel Iflavirus in Wild and Cultivated Acheta domesticus. Viruses 2021, 13, 364. https://doi.org/10.3390/v13030364
de Miranda JR, Granberg F, Onorati P, Jansson A, Berggren Å. Virus Prospecting in Crickets—Discovery and Strain Divergence of a Novel Iflavirus in Wild and Cultivated Acheta domesticus. Viruses. 2021; 13(3):364. https://doi.org/10.3390/v13030364
Chicago/Turabian Stylede Miranda, Joachim R., Fredrik Granberg, Piero Onorati, Anna Jansson, and Åsa Berggren. 2021. "Virus Prospecting in Crickets—Discovery and Strain Divergence of a Novel Iflavirus in Wild and Cultivated Acheta domesticus" Viruses 13, no. 3: 364. https://doi.org/10.3390/v13030364
APA Stylede Miranda, J. R., Granberg, F., Onorati, P., Jansson, A., & Berggren, Å. (2021). Virus Prospecting in Crickets—Discovery and Strain Divergence of a Novel Iflavirus in Wild and Cultivated Acheta domesticus. Viruses, 13(3), 364. https://doi.org/10.3390/v13030364