
mRNA-lncRNA Co-Expression Network Analysis Reveals the Role of lncRNAs in Immune Dysfunction during Severe SARS-CoV-2 Infection




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Acquisition of Transcriptome Dataset
2.2. Read Mapping and Differential Expression Analysis
2.3. Co-Expression Network Analysis
2.4. Gene Ontology and Pathway Enrichment Analysis
3. Results
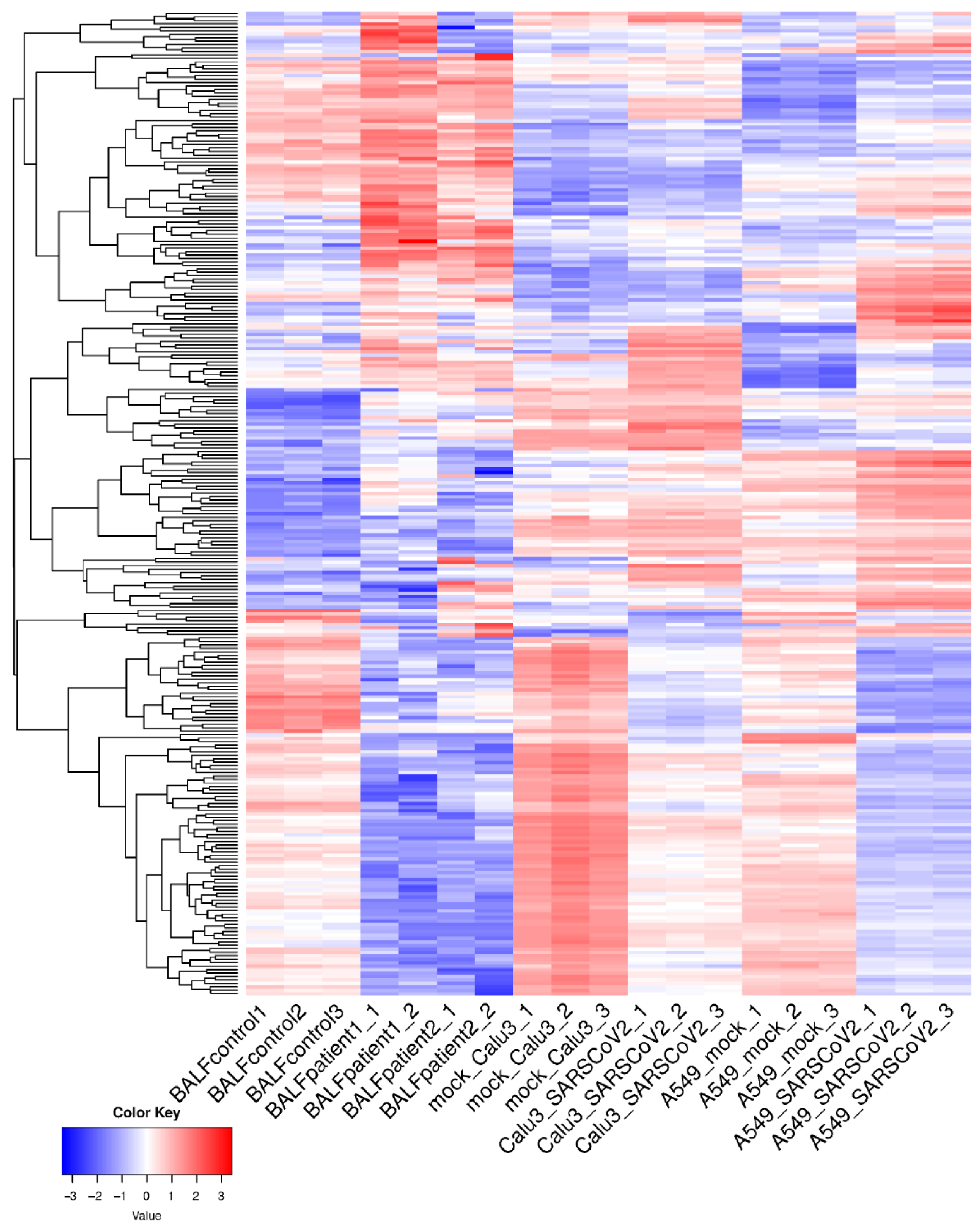
3.1. Identification of Common DE mRNAs and lncRNAs in Response to SARS-CoV-2 Infection of the Lungs
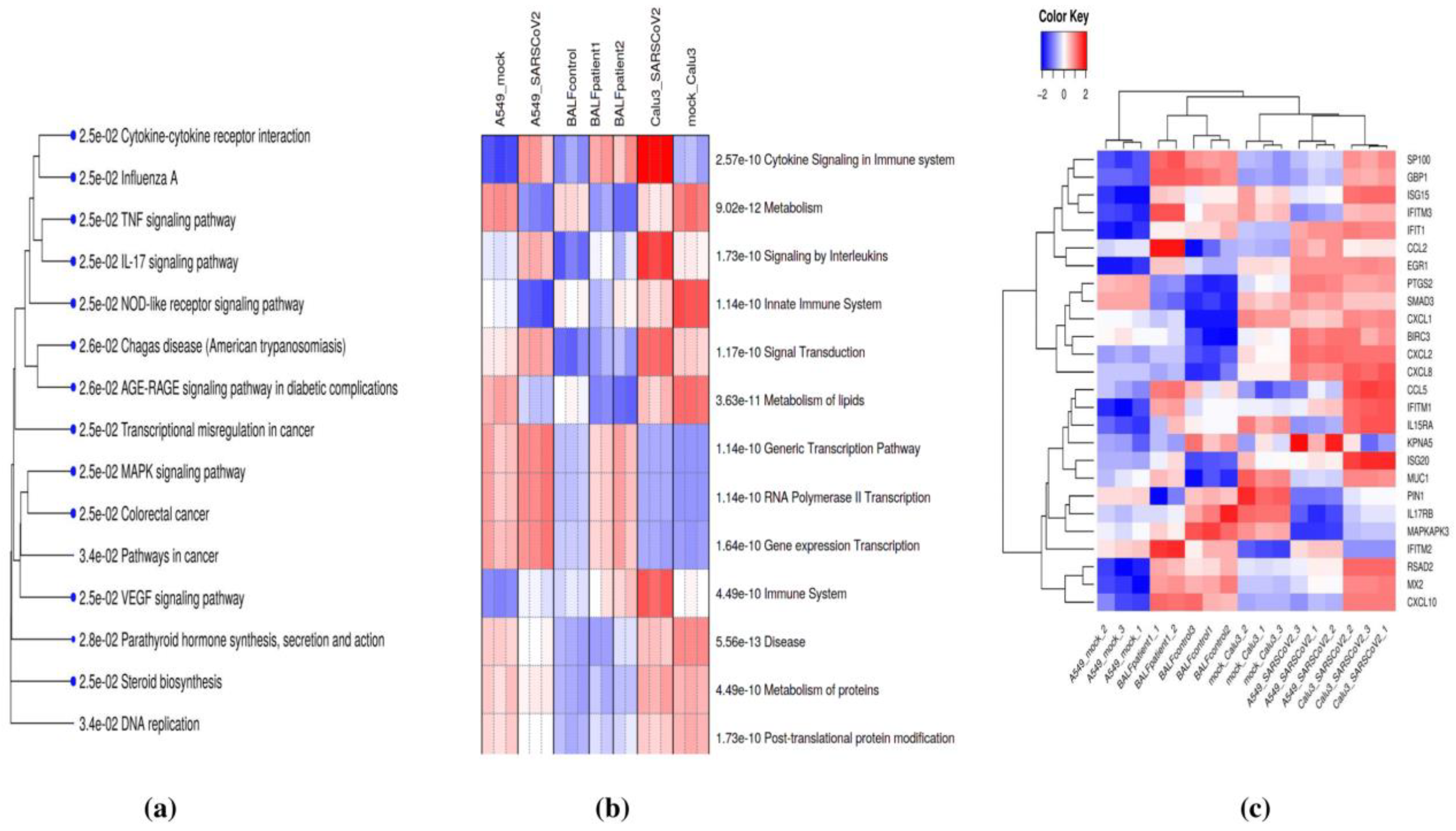
3.2. Pathway Enrichment Analysis of Common DE Genes Highlight Potential Roles in Cytokine Signaling
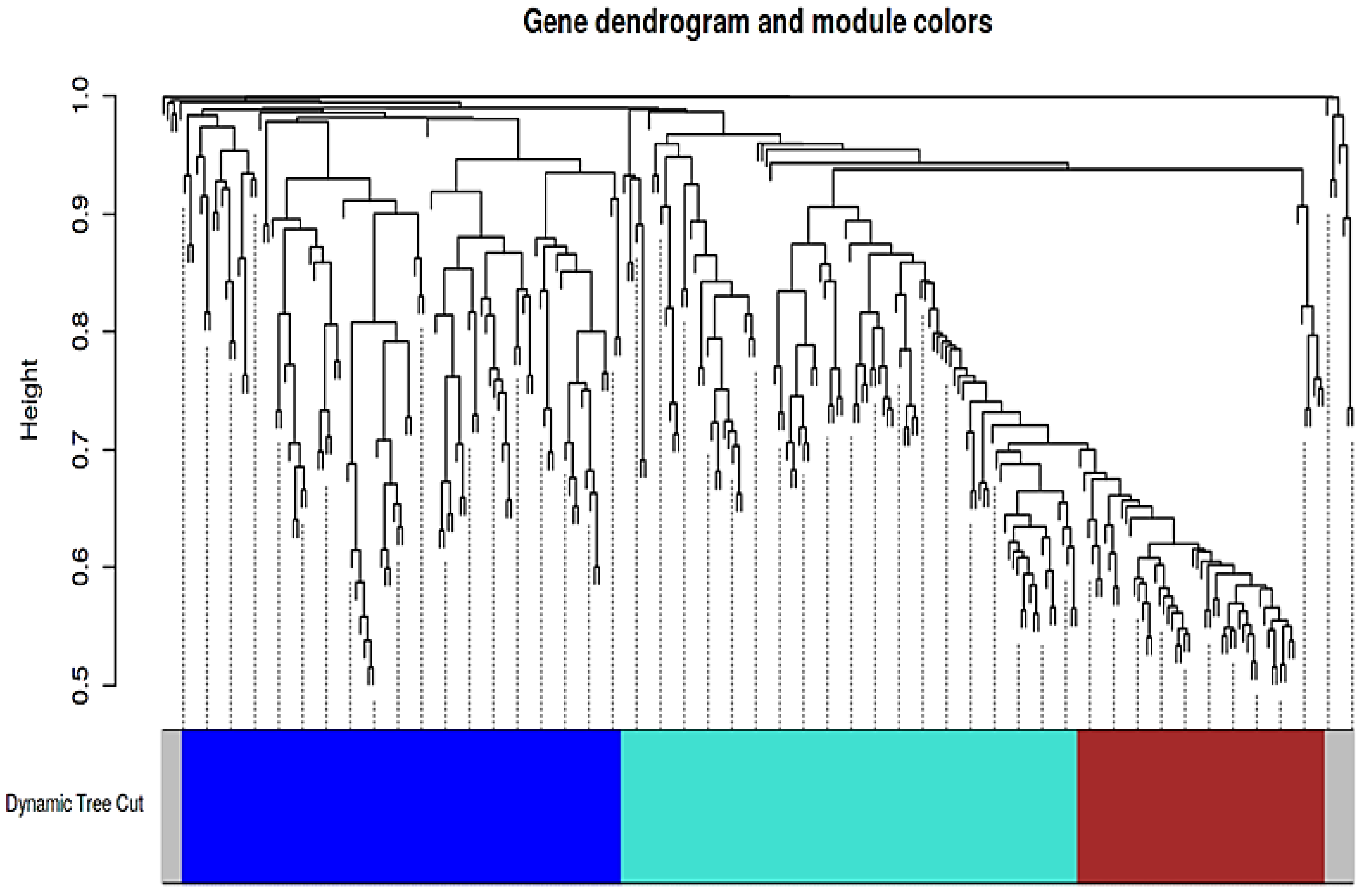
3.3. Co-Expression Analysis and Identification of a Key Module Associated with Cytokine Signaling
3.4. Analysis of Hub Nodes from lncRNA-mRNA Co-Expression Networks Reveals the Potential Involvement of lncRNAs in Cytokine Signaling
3.5. Expression Profiling of lncRNAs and Their Interactors in the SARS and MERS-Infected Calu3 Cell Line
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Lai, C.C.; Shih, T.P.; Ko, W.C.; Tang, H.J.; Hsueh, P.R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int. J. Antimicrob. Agents 2020, 55, 105924. [Google Scholar] [CrossRef]
- Dong, Y.; Dai, T.; Wei, Y.; Zhang, L.; Zheng, M.; Zhou, F. A systematic review of SARS-CoV-2 vaccine candidates. Signal Transduct. Target. Ther. 2020, 5, 237. [Google Scholar] [CrossRef] [PubMed]
- Tworowski, D.; Gorohovski, A.; Mukherjee, S.; Carmi, G.; Levy, E.; Detroja, R.; Mukherjee, S.B.; Frenkel-Morgenstern, M. COVID19 Drug Repository: Text-mining the literature in search of putative COVID19 therapeutics. Nucleic Acids Res. 2020. [Google Scholar] [CrossRef]
- Mukherjee, S.; Tworowski, D.; Detroja, R.; Mukherjee, S.B.; Frenkel-Morgenstern, M. Immunoinformatics and structural analysis for identification of immunodominant epitopes in SARS-CoV-2 as potential vaccine targets. Vaccines 2020, 8, 290. [Google Scholar] [CrossRef] [PubMed]
- Channappanavar, R.; Perlman, S. Pathogenic human coronavirus infections: Causes and consequences of cytokine storm and immunopathology. Semin. Immunopathol. 2017, 39, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Fan, E.; Beitler, J.R.; Brochard, L.; Calfee, C.S.; Ferguson, N.D.; Slutsky, A.S.; Brodie, D. COVID-19-associated acute respiratory distress syndrome: Is a different approach to management warranted? Lancet Respir. Med. 2020, 8, 816–821. [Google Scholar] [CrossRef]
- Berlin, D.A.; Gulick, R.M.; Martinez, F.J. Severe Covid-19. N. Engl. J. Med. 2020, 383, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Ortiz-Prado, E.; Simbaña-Rivera, K.; Gómez-Barreno, L.; Rubio-Neira, M.; Guaman, L.P.; Kyriakidis, N.C.; Muslin, C.; Jaramillo, A.M.G.; Barba-Ostria, C.; Cevallos-Robalino, D.; et al. Clinical, molecular, and epidemiological characterization of the SARS-CoV-2 virus and the Coronavirus Disease 2019 (COVID-19), a comprehensive literature review. Diagn. Microbiol. Infect. Dis. 2020, 98, 115094. [Google Scholar] [CrossRef] [PubMed]
- Rockx, B.; Kuiken, T.; Herfst, S.; Bestebroer, T.; Lamers, M.M.; Munnink, B.B.O.; De Meulder, D.; Van Amerongen, G.; Van Den Brand, J.; Okba, N.M.A.; et al. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science 2020, 368, 1012–1015. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Vaninov, N. In the eye of the COVID-19 cytokine storm. Nat. Rev. Immunol. 2020, 20, 277. [Google Scholar] [CrossRef] [PubMed]
- Mangalmurti, N.; Hunter, C.A. Cytokine Storms: Understanding COVID-19. Immunity 2020, 53, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Henderson, L.A.; Canna, S.W.; Schulert, G.S.; Volpi, S.; Lee, P.Y.; Kernan, K.F.; Caricchio, R.; Mahmud, S.; Hazen, M.M.; Halyabar, O.; et al. On the Alert for Cytokine Storm: Immunopathology in COVID-19. Arthritis Rheumatol. 2020, 72, 1059–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Coperchini, F.; Chiovato, L.; Croce, L.; Magri, F.; Rotondi, M. The cytokine storm in COVID-19: An overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev. 2020, 53, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Fara, A.; Mitrev, Z.; Rosalia, R.A.; Assas, B.M. Cytokine storm and COVID-19: A chronicle of pro-inflammatory cytokines: Cytokine storm: The elements of rage! Open Biol. 2020, 10, 200160. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.; Davidson, S.M.; Yellon, D.M. The cytokine storm of COVID-19: A spotlight on prevention and protection. Expert Opin. Ther. Targets 2020, 24, 723–730. [Google Scholar] [CrossRef]
- Chen, L.Y.C.; Hoiland, R.L.; Stukas, S.; Wellington, C.L.; Sekhon, M.S. Confronting the controversy: Interleukin-6 and the COVID-19 cytokine storm syndrome. Eur. Respir. J. 2020, 56, 2003006. [Google Scholar] [CrossRef]
- Chen, L.D.; Zhang, Z.Y.; Wei, X.J.; Cai, Y.Q.; Yao, W.Z.; Wang, M.H.; Huang, Q.F.; Zhang, X. Bin Association between cytokine profiles and lung injury in COVID-19 pneumonia. Respir. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Ganeshan, K.; Chawla, A. Metabolic regulation of immune responses. Annu. Rev. Immunol. 2014, 32, 609–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs: Insights into functions. Nat. Rev. Genet. 2009, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rubio, A.; Ghosh, S. Disease-associated SNPs in inflammation-related lncRNAs. Front. Immunol. 2019, 10, 420. [Google Scholar] [CrossRef] [Green Version]
- Taft, R.J.; Pang, K.C.; Mercer, T.R.; Dinger, M.; Mattick, J.S. Non-coding RNAs: Regulators of disease. J. Pathol. 2010, 220, 126–139. [Google Scholar] [CrossRef] [PubMed]
- Geng, H.; Tan, X. Di Functional diversity of long non-coding RNAs in immune regulation. Genes Dis. 2016, 3, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Ao, L.; Yang, J. Long non-coding RNAs in diseases related to inflammation and immunity. Ann. Transl. Med. 2019. [Google Scholar] [CrossRef] [PubMed]
- Heward, J.A.; Lindsay, M.A. Long non-coding RNAs in the regulation of the immune response. Trends Immunol. 2014, 35, 408–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Liu, Y.; Cao, L.; Wang, D.; Guo, M.; Jiang, A.; Guo, D.; Hu, W.; Yang, J.; Tang, Z.; et al. Transcriptomic characteristics of bronchoalveolar lavage fluid and peripheral blood mononuclear cells in COVID-19 patients. Emerg. Microbes Infect. 2020, 9, 761–770. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Chin, C.H.; Chen, S.H.; Wu, H.H.; Ho, C.W.; Ko, M.T.; Lin, C.Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8, S11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, B.; Koner, D.; Karasik, D.; Saha, N. Genome-wide identification of novel long non-coding RNAs and their possible roles in hypoxic zebrafish brain. Genomics 2021, 113, 29–43. [Google Scholar] [CrossRef]
- Ge, S.X.; Jung, D.; Jung, D.; Yao, R. ShinyGO: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019. [Google Scholar] [CrossRef]
- Kim, S.Y.; Volsky, D.J. PAGE: Parametric analysis of gene set enrichment. BMC Bioinform. 2005, 6, 144. [Google Scholar] [CrossRef] [Green Version]
- Blanco-Melo, D.; Nilsson-Payant, B.E.; Liu, W.C.; Uhl, S.; Hoagland, D.; Møller, R.; Jordan, T.X.; Oishi, K.; Panis, M.; Sachs, D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181, 1036–1045. [Google Scholar] [CrossRef]
- Prieto, C.; Risueño, A.; Fontanillo, C.; De Las Rivas, J. Human gene coexpression landscape: Confident network derived from tissue transcriptomic profiles. PLoS ONE 2008, 3, e3911. [Google Scholar] [CrossRef] [Green Version]
- Hojyo, S.; Uchida, M.; Tanaka, K.; Hasebe, R.; Tanaka, Y.; Murakami, M.; Hirano, T. How COVID-19 induces cytokine storm with high mortality. Inflamm. Regen. 2020, 40, 37. [Google Scholar] [CrossRef]
- Leisman, D.E.; Ronner, L.; Pinotti, R.; Taylor, M.D.; Sinha, P.; Calfee, C.S.; Hirayama, A.V.; Mastroiani, F.; Turtle, C.J.; Harhay, M.O.; et al. Cytokine elevation in severe and critical COVID-19: A rapid systematic review, meta-analysis, and comparison with other inflammatory syndromes. Lancet Respir. Med. 2020, 8, 1233–1244. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Hariharan, A.; Hakeem, A.R.; Radhakrishnan, S.; Reddy, M.S.; Rela, M. The Role and Therapeutic Potential of NF-kappa-B Pathway in Severe COVID-19 Patients. Inflammopharmacology 2021, 29, 91–100. [Google Scholar] [CrossRef]
- Shibabaw, T. Inflammatory cytokine: Il-17a signaling pathway in patients present with covid-19 and current treatment strategy. J. Inflamm. Res. 2020, 13, 673–680. [Google Scholar] [CrossRef]
- Tobinick, E. TNF-α inhibition for potential therapeutic modulation of SARS coronavirus infection. Curr. Med. Res. Opin. 2004, 20, 39–40. [Google Scholar] [CrossRef] [PubMed]
- Karki, R.; Sharma, B.R.; Tuladhar, S.; Williams, E.P.; Zalduondo, L.; Samir, P.; Zheng, M.; Sundaram, B.; Banoth, B.; Malireddi, R.S.; et al. COVID-19 cytokines and the hyperactive immune response: Synergism of TNF-α and IFN-γ in triggering inflammation, tissue damage, and death. bioRxiv 2020. [Google Scholar] [CrossRef]
- Herter, E.K.; Li, D.; Toma, M.A.; Vij, M.; Li, X.; Visscher, D.; Wang, A.; Chu, T.; Sommar, P.; Blomqvist, L.; et al. WAKMAR2, a Long Noncoding RNA Downregulated in Human Chronic Wounds, Modulates Keratinocyte Motility and Production of Inflammatory Chemokines. J. Investig. Dermatol. 2019, 139, 1373–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carnero, E.; Barriocanal, M.; Prior, C.; Pablo Unfried, J.; Segura, V.; Guruceaga, E.; Enguita, M.; Smerdou, C.; Gastaminza, P.; Fortes, P. Long noncoding RNA EGOT negatively affects the antiviral response and favors HCV replication. EMBO Rep. 2016, 17, 1013–1028. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mukherjee, S.; Banerjee, B.; Karasik, D.; Frenkel-Morgenstern, M. mRNA-lncRNA Co-Expression Network Analysis Reveals the Role of lncRNAs in Immune Dysfunction during Severe SARS-CoV-2 Infection. Viruses 2021, 13, 402. https://doi.org/10.3390/v13030402
Mukherjee S, Banerjee B, Karasik D, Frenkel-Morgenstern M. mRNA-lncRNA Co-Expression Network Analysis Reveals the Role of lncRNAs in Immune Dysfunction during Severe SARS-CoV-2 Infection. Viruses. 2021; 13(3):402. https://doi.org/10.3390/v13030402
Chicago/Turabian StyleMukherjee, Sumit, Bodhisattwa Banerjee, David Karasik, and Milana Frenkel-Morgenstern. 2021. "mRNA-lncRNA Co-Expression Network Analysis Reveals the Role of lncRNAs in Immune Dysfunction during Severe SARS-CoV-2 Infection" Viruses 13, no. 3: 402. https://doi.org/10.3390/v13030402
APA StyleMukherjee, S., Banerjee, B., Karasik, D., & Frenkel-Morgenstern, M. (2021). mRNA-lncRNA Co-Expression Network Analysis Reveals the Role of lncRNAs in Immune Dysfunction during Severe SARS-CoV-2 Infection. Viruses, 13(3), 402. https://doi.org/10.3390/v13030402