
Picornaviruses: A View from 3A


Abstract
:1. Picornaviruses
2. Key Features of Picornavirus Genomes
3. Key Features of the Picornavirus Replication-Cycle
4. Picornavirus 3A Proteins
5. Viral Binding Partner Proteins
5.1. Dimerization of PV 3A and 3AB
5.2. Interactions between PV 3A/3AB and the P2 Proteins (2B, 2C and 2BC)
5.3. Interactions between PV 3A/3AB and 3Dpol, 3CDpro and 2Apro
5.4. Binding Partners of AiV 3A
6. Inhibition of Protein Secretion
7. Membrane Interactions
8. Host-Range Determinant
9. Nucleic Acid Chaperone Activity
10. Interactions with Cellular Proteins
10.1. LIS1
10.2. Dynactin-3
10.3. Vimentin
11. Evasion of the Cellular Antiviral Response
11.1. ATP1B3
11.2. RIG-I, MDA5 and MAVS
11.3. DDX56
11.4. G3BP1
11.5. RNA Interference
12. The Viral Replication Complex and vRNA Replication
13. Uridylylation of VPg (3B)
14. Formation of Replication Organelles
15. Recruitment of PI4K to Replication Organelles
16. Recruitment of PI4KB by Subversion of GBF1/ARF1
16.1. The Case for 3A
16.2. The Case for 3CDpro
17. 3A and the Great Escapes
18. Recruitment of PI4KB by Subversion of ACBD3
19. Lipid Droplets
20. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Zell, R. Picornaviridae—the ever-growing virus family. Arch. Virol. 2018, 163, 299–317. [Google Scholar] [CrossRef]
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Knight-Jones, T.; Rushton, J. The economic impacts of foot and mouth disease—What are they, how big are they and where do they occur? Prev. Veter Med. 2013, 112, 161–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexandersen, S.; Knowles, N.J.; Belsham, G.J.; Dekker, A.; Nfon, C.; Zhang, Z.; Koenen, F. Picornaviruses. In Diseases of Swine; Wiley: Hoboken, NJ, USA, 2019; pp. 641–684. [Google Scholar]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef]
- Belsham, G.J. Divergent picornavirus IRES elements. Virus Res. 2009, 139, 183–192. [Google Scholar] [CrossRef]
- Lange, J.; Groth, M.; Fichtner, D.; Granzow, H.; Keller, B.; Walther, M.; Platzer, M.; Sauerbrei, A.; Zell, R. Virus isolate from carp: Genetic characterization reveals a novel picornavirus with two aphthovirus 2A-like sequences. J. Gen. Virol. 2014, 95, 80–90. [Google Scholar] [CrossRef] [Green Version]
- Reuter, G.; Boros, Á.; Földvári, G.; Szekeres, S.; Mátics, R.; Kapusinszky, B.; Delwart, E.; Pankovics, P. Dicipivirus (family Picornaviridae) in wild Northern white-breasted hedgehog (Erinaceus roumanicus). Arch. Virol. 2018, 163, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Cheng, A.; Wang, M.; Jia, R.; Sun, K.; Pan, K.; Yang, Q.; Wu, Y.; Zhu, D.; Chen, S.; et al. Structures and Corresponding Functions of Five Types of Picornaviral 2A Proteins. Front. Microbiol. 2017, 8, 1373. [Google Scholar] [CrossRef] [Green Version]
- Ambros, V.; Baltimore, D. Protein is linked to the 5’ end of poliovirus RNA by a phosphodiester linkage to tyrosine. J. Biol. Chem. 1978, 253, 5263–5266. [Google Scholar] [CrossRef]
- Flanegan, J.B.; Petterson, R.F.; Ambros, V.; Hewlett, N.J.; Baltimore, D. Covalent linkage of a protein to a defined nucleotide sequence at the 5’-terminus of virion and replicative intermediate RNAs of poliovirus. In Proceedings of the Proceedings of the National Academy of Sciences; Proc. Natl. Acad. Sci. USA 1977, 74, 961–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.F.; Nomoto, A.; Detjen, B.M.; Wimmer, E. A protein covalently linked to poliovirus genome RNA. Proc. Natl. Acad. Sci. USA 1977, 74, 59–63. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.V.; Van Boom, J.H.; Filippov, D.V.; Wimmer, E. Protein-primed RNA synthesis by purified poliovirus RNA polymerase. Nat. Cell Biol. 1998, 393, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, P.G.; Harris, T.J.; Nomoto, A.; Wimmer, E. O4-(5’-uridylyl)tyrosine is the bond between the genome-linked protein and the RNA of poliovirus. Proc. Natl. Acad. Sci. USA 1978, 75, 4868–4872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andino, R.; Rieckhof, G.; Achacoso, P.; Baltimore, D. Poliovirus RNA synthesis utilizes an RNP complex formed around the 5′-end of viral RNA. EMBO J. 1993, 12, 3587–3598. [Google Scholar] [CrossRef]
- Barton, D.J.; O’Donnell, B.J.; Flanegan, J.B. 5′ cloverleaf in poliovirus RNA is a cis-acting replication element required for negative-strand synthesis. EMBO J. 2001, 20, 1439–1448. [Google Scholar] [CrossRef]
- Kloc, A.; Segundo, F.D.-S.; Schafer, E.A.; Rai, D.K.; Kenney, M.; Santos, T.D.L.; Rieder, E. Foot-and-mouth disease virus 5’-terminal S fragment is required for replication and modulation of the innate immune response in host cells. Virology 2017, 512, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Salas, E.; Francisco-Velilla, R.; Fernandez-Chamorro, J.; Lozano, G.; Diaz-Toledano, R. Picornavirus IRES elements: RNA structure and host protein interactions. Virus Res. 2015, 206, 62–73. [Google Scholar] [CrossRef]
- Vogt, D.A.; Andino, R. An RNA Element at the 5′-End of the Poliovirus Genome Functions as a General Promoter for RNA Synthesis. PLoS Pathog. 2010, 6, e1000936. [Google Scholar] [CrossRef] [Green Version]
- Herold, J.; Andino, R. Poliovirus RNA Replication Requires Genome Circularization through a Protein–Protein Bridge. Mol. Cell 2001, 7, 581–591. [Google Scholar] [CrossRef]
- Ogram, S.A.; Flanegan, J.B. Non-template functions of viral RNA in picornavirus replication. Curr. Opin. Virol. 2011, 1, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Sarnow, P. Role of 3’-end sequences in infectivity of poliovirus transcripts made in vitro. J. Virol. 1989, 63, 467–470. [Google Scholar] [CrossRef] [Green Version]
- Serrano, P. The 3’ end of the foot-and-mouth disease virus genome establishes two distinct long-range RNA-RNA interactions with the 5’ end region. J. Gen. Virol. 2006, 87, 3013–3022. [Google Scholar] [CrossRef]
- Van Ooij, M.J.M. Polyadenylation of genomic RNA and initiation of antigenomic RNA in a positive-strand RNA virus are controlled by the same cis-element. Nucleic Acids Res. 2006, 34, 2953–2965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Bakkers, J.M.J.E.; Galama, J.M.D.; Slot, H.J.B.; Pilipenko, E.V.; Agol, V.I.; Melchers, W.J.G. Structural requirements of the higher order RNA kissing element in the enteroviral 3’UTR. Nucleic Acids Res. 1999, 27, 485–490. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, I.; Chaudhry, Y.; Richardson, A.; Meredith, J.; Almond, J.W.; Barclay, W.; Evans, D.J. Identification of a cis-Acting Replication Element within the Poliovirus Coding Region. J. Virol. 2000, 74, 4590–4600. [Google Scholar] [CrossRef]
- Mason, P.W.; Bezborodova, S.V.; Henry, T.M. Identification and Characterization of a cis-Acting Replication Element (cre) Adjacent to the Internal Ribosome Entry Site of Foot-and-Mouth Disease Virus. J. Virol. 2002, 76, 9686–9694. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKnight, K.L.; Lemon, S.M. The rhinovirus type 14 genome contains an internally located RNA structure that is required for viral replication. RNA 1998, 4, 1569–1584. [Google Scholar] [CrossRef] [Green Version]
- Nayak, A.; Goodfellow, I.G.; Belsham, G.J. Factors Required for the Uridylylation of the Foot-and-Mouth Disease Virus 3B1, 3B2, and 3B3 Peptides by the RNA-Dependent RNA Polymerase (3Dpol) In Vitro. J. Virol. 2005, 79, 7698–7706. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.V.; Rieder, E.; Kim, D.W.; Van Boom, J.H.; Wimmer, E. Identification of an RNA Hairpin in Poliovirus RNA That Serves as the Primary Template in the In Vitro Uridylylation of VPg. J. Virol. 2000, 74, 10359–10370. [Google Scholar] [CrossRef] [Green Version]
- Belsham, G.J.; Bostock, C.J. Studies on the Infectivity of Foot-and-Mouth Disease Virus RNA using Microinjection. J. Gen. Virol. 1988, 69, 265–274. [Google Scholar] [CrossRef]
- Holland, J.J.; Bassett, D.W. Evidence for cytoplasmic replication of poliovirus ribonucleic acid. Virology 1964, 23, 164–172. [Google Scholar] [CrossRef]
- Tuthill, T.J.; Bubeck, D.; Rowlands, D.J.; Hogle, J.M. Characterization of Early Steps in the Poliovirus Infection Process: Receptor-Decorated Liposomes Induce Conversion of the Virus to Membrane-Anchored Entry-Intermediate Particles. J. Virol. 2006, 80, 172–180. [Google Scholar] [CrossRef] [Green Version]
- Bergamini, G.; Preiss, T.; Hentze, M.W. Picornavirus IRESes and the poly(A) tail jointly promote cap-independent translation in a mammalian cell-free system. RNA 2000, 6, 1781–1790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Nuñez, S.; Gismondi, M.I.; König, G.; Berinstein, A.; Taboga, O.; Rieder, E.; Martínez-Salas, E.; Carrillo, E. Enhanced IRES activity by the 3′UTR element determines the virulence of FMDV isolates. Virology 2014, 448, 303–313. [Google Scholar] [CrossRef] [Green Version]
- De Quinto, S.L. IRES-driven translation is stimulated separately by the FMDV 3’-NCR and poly(A) sequences. Nucleic Acids Res. 2002, 30, 4398–4405. [Google Scholar] [CrossRef]
- Simoes, E.A.; Sarnow, P. An RNA hairpin at the extreme 5’ end of the poliovirus RNA genome modulates viral translation in human cells. J. Virol. 1991, 65, 913–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, B.E.; Sangar, D.V. Processing and Assembly of Foot-and-Mouth Disease Virus Proteins Using Subgenomic RNA. J. Gen. Virol. 1988, 69, 2313–2325. [Google Scholar] [CrossRef] [PubMed]
- Kräusslich, H.-G.; Nicklin, M.J.; Lee, C.-K.; Wimmer, E. Polyprotein processing in picornavirus replication. Biochimestry 1988, 70, 119–130. [Google Scholar] [CrossRef]
- Lawson, M.A.; Semler, B.L. Picornavirus Protein Processing—Enzymes, Substrates, and Genetic Regulation. Curr. Topics Microbiol. Immunol. 1990, 161, 49–87. [Google Scholar] [CrossRef]
- Pallansch, M.A.; Kew, O.M.; Semler, B.L.; Omilianowski, D.R.; Anderson, C.W.; Wimmer, E.; Rueckert, R.R. Protein processing map of poliovirus. J. Virol. 1984, 49, 873–880. [Google Scholar] [CrossRef] [Green Version]
- Strebel, K.; Beck, E. A second protease of foot-and-mouth disease virus. J. Virol. 1986, 58, 893–899. [Google Scholar] [CrossRef] [Green Version]
- Parks, G.D.; Duke, G.M.; Palmenberg, A.C. Encephalomyocarditis virus 3C protease: Efficient cell-free expression from clones which link viral 5’ noncoding sequences to the P3 region. J. Virol. 1986, 60, 376–384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, J.; Taniguchi, K. Aichi Virus 2A Protein Is Involved in Viral RNA Replication. J. Virol. 2008, 82, 9765–9769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmenberg, A.C. Proteolytic Processing of Picornaviral Polyprotein. Annu. Rev. Microbiol. 1990, 44, 603–623. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, J.; Ishikawa, K.; Taniguchi, K. 3CD, but not 3C, cleaves the VP1/2A site efficiently during Aichi virus polyprotein processing through interaction with 2A. Virus Res. 2012, 163, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Donnelly, M.L.L.; Luke, G.; Mehrotra, A.; Li, X.; Hughes, L.E.; Gani, D.; Ryan, M.D. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: A putative ribosomal ‘skip’. J. Gen. Virol. 2001, 82, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Palmenberg, A.C.; Parks, G.D.; Hall, D.J.; Ingraham, R.H.; Seng, T.W.; Pallai, P.V. Proteolytic processing of the cardioviral P2 region: Primary 2A/2B cleavage in clone-derived precursors. Virology 1992, 190, 754–762. [Google Scholar] [CrossRef]
- Belsham, G.J.; Jackson, R.J. Translation initiation on picornavirus RNA. In Translational Control of Gene Expression; Monograph 39; Sonenberg, N., Hershey, J.W.B., Mathews, M.B., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2000; pp. 869–900. [Google Scholar]
- Paul, A.V.; Wimmer, E. Initiation of protein-primed picornavirus RNA synthesis. Virus Res. 2015, 206, 12–26. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, D.; Yamashita, S.; Tomita, K. Molecular insights into replication initiation by Qβ replicase using ribosomal protein S1. Nucleic Acids Res. 2014, 42, 10809–10822. [Google Scholar] [CrossRef] [Green Version]
- Pilipenko, E.V.; Poperechny, K.V.; Maslova, S.V.; Melchers, W.J.; Slot, H.J.; Agol, V.I. Cis-element, oriR, involved in the initiation of (-) strand poliovirus RNA: A quasi-globular multi-domain RNA structure maintained by tertiary (‘kissing’) in-teractions. EMBO J. 1996, 15, 5428–5436. [Google Scholar] [CrossRef] [Green Version]
- Flanegan, J.B.; Baltimore, D. Poliovirus-specific primer-dependent RNA polymerase able to copy poly(A). Proc. Natl. Acad. Sci. USA 1977, 74, 3677–3680. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Victoria, J.; Simmonds, P.; Wang, C.; Shafer, R.W.; Nims, R.; Nielsen, O.; Delwart, E. A Highly Divergent Picornavirus in a Marine Mammal. J. Virol. 2007, 82, 311–320. [Google Scholar] [CrossRef] [Green Version]
- Forss, S.; Schaller, H. A tandem repeat gene in a picornavirus. Nucleic Acids Res. 1982, 10, 6441–6450. [Google Scholar] [CrossRef] [PubMed]
- Forss, S.; Strebel, K.; Beck, E.; Schaller, H. Nucleotide sequence and genome organization of foot-and-mouth disease virus. Nucleic Acids Res. 1984, 12, 6587–6601. [Google Scholar] [CrossRef] [Green Version]
- King, A.M.; Sangar, D.V.; Harris, T.J.; Brown, F. Heterogeneity of the genome-linked protein of foot-and-mouth disease virus. J. Virol. 1980, 34, 627–634. [Google Scholar] [CrossRef] [Green Version]
- Logan, G.; Newman, J.; Wright, C.F.; Lasecka-Dykes, L.; Haydon, D.T.; Cottam, E.M.; Tuthill, T.J. Deep Sequencing of Foot-and-Mouth Disease Virus Reveals RNA Sequences Involved in Genome Packaging. J. Virol. 2017, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, H.-C.; Liu, Y.; Wang, C.; Strauss, M.; Rehage, N.; Chen, Y.-H.; Altan-Bonnet, N.; Hogle, J.; Wimmer, E.; Mueller, S.; et al. An Interaction between Glutathione and the Capsid Is Required for the Morphogenesis of C-Cluster Enteroviruses. PLoS Pathog. 2014, 10, e1004052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, J.; Nagashima, S.; Taniguchi, K. Aichi Virus Leader Protein Is Involved in Viral RNA Replication and Encapsidation. J. Virol. 2003, 77, 10799–10807. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, J.; Taniguchi, K. The 5′-End Sequence of the Genome of Aichi Virus, a Picornavirus, Contains an Element Critical for Viral RNA Encapsidation. J. Virol. 2003, 77, 3542–3548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Jiang, P.; Sand, C.; Paul, A.V.; Wimmer, E. Alanine Scanning of Poliovirus 2CATPaseReveals New Genetic Evidence that Capsid Protein/2CATPaseInteractions Are Essential for Morphogenesis. J. Virol. 2012, 86, 9964–9975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chow, M.; Newman, J.F.E.; Filman, D.; Hogle, J.M.; Rowlands, D.J.; Brown, F. Myristylation of picornavirus capsid protein VP4 and its structural significance. Nat. Cell Biol. 1987, 327, 482–486. [Google Scholar] [CrossRef]
- Ramljak, I.C.; Stanger, J.; Real-Hohn, A.; Dreier, D.; Wimmer, L.; Redlberger-Fritz, M.; Fischl, W.; Klingel, K.; Mihovilovic, M.D.; Blaas, D.; et al. Cellular N-myristoyltransferases play a crucial picornavirus genus-specific role in viral assembly, virion maturation, and infectivity. PLoS Pathog. 2018, 14, e1007203. [Google Scholar] [CrossRef]
- Paul, A.V.; Schultz, A.; Pincus, S.E.; Oroszlan, S.; Wimmer, E. Capsid protein VP4 of poliovirus is N-myristoylated. Proc. Natl. Acad. Sci. USA 1987, 84, 7827–7831. [Google Scholar] [CrossRef] [Green Version]
- Hogle, J.M. Poliovirus Cell Entry: Common Structural Themes in Viral Cell Entry Pathways. Annu. Rev. Microbiol. 2002, 56, 677–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panjwani, A.; Strauss, M.; Gold, S.; Wenham, H.; Jackson, T.; Chou, J.J.; Rowlands, D.J.; Stonehouse, N.J.; Hogle, J.M.; Tuthill, T.J. Capsid Protein VP4 of Human Rhinovirus Induces Membrane Permeability by the Formation of a Size-Selective Multimeric Pore. PLoS Pathog. 2014, 10, e1004294. [Google Scholar] [CrossRef] [PubMed]
- Stanway, G.; Kalkkinen, N.; Roivainen, M.; Ghazi, F.; Khan, M.; Smyth, M.; Meurman, O.; Hyypiä, T. Molecular and biological characteristics of echovirus 22, a representative of a new picornavirus group. J. Virol. 1994, 68, 8232–8238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesar, M.; Jia, X.-Y.; Summers, D.F.; Ehrenfeld, E. Analysis of a Potential Myristoylation Site in Hepatitis A Virus Capsid Protein VP4. Virology 1993, 194, 616–626. [Google Scholar] [CrossRef] [PubMed]
- Basavappa, R.; Filman, D.J.; Syed, R.; Flore, O.; Icenogle, J.P.; Hogle, J.M. Role and mechanism of the maturation cleavage of VP0 in poliovirus assembly: Structure of the empty capsid assembly intermediate at 2.9 Å resolution. Protein Sci. 1994, 3, 1651–1669. [Google Scholar] [CrossRef] [Green Version]
- Stanway, G.; Hyypia, T. Parechoviruses. J. Virol. 1999, 73, 5249–5254. [Google Scholar] [CrossRef] [Green Version]
- Yamashita, T.; Sakae, K.; Tsuzuki, H.; Suzuki, Y.; Ishikawa, N.; Takeda, N.; Miyamura, T.; Yamazaki, S. Complete Nucleotide Sequence and Genetic Organization of Aichi Virus, a Distinct Member of the PicornaviridaeAssociated with Acute Gastroenteritis in Humans. J. Virol. 1998, 72, 8408–8412. [Google Scholar] [CrossRef] [Green Version]
- Curry, S.; Fry, E.; Blakemore, W.; Abu-Ghazaleh, R.; Jackson, T.; King, A.; Lea, S.; Newman, J.; Stuart, D. Dissecting the roles of VP0 cleavage and RNA packaging in picornavirus capsid stabilization: The structure of empty capsids of foot-and-mouth disease virus. J. Virol. 1997, 71, 9743–9752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gullberg, M.; Muszynski, B.; Organtini, L.J.; Ashley, R.E.; Hafenstein, S.L.; Belsham, G.J.; Polacek, C. Assembly and characterization of foot-and-mouth disease virus empty capsid particles expressed within mammalian cells. J. Gen. Virol. 2013, 94, 1769–1779. [Google Scholar] [CrossRef] [PubMed]
- Robinson, S.M.; Tsueng, G.; Sin, J.; Mangale, V.; Rahawi, S.; McIntyre, L.L.; Williams, W.; Kha, N.; Cruz, C.; Hancock, B.M.; et al. Coxsackievirus B Exits the Host Cell in Shed Microvesicles Displaying Autophagosomal Markers. PLoS Pathog. 2014, 10, e1004045. [Google Scholar] [CrossRef]
- Taylor, M.P.; Burgon, T.B.; Kirkegaard, K.; Jackson, W.T. Role of Microtubules in Extracellular Release of Poliovirus. J. Virol. 2009, 83, 6599–6609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Grein, S.G.; Defourny, K.A.Y.; Rabouw, H.H.; Galiveti, C.R.; Langereis, M.A.; Wauben, M.H.M.; Arkesteijn, G.J.A.; Van Kuppeveld, F.J.M.; Hoen, E.N.M.N. ‘T Picornavirus infection induces temporal release of multiple extracellular vesicle subsets that differ in molecular composition and infectious potential. PLoS Pathog. 2019, 15, e1007594. [Google Scholar] [CrossRef] [Green Version]
- Doedens, J.; Kirkegaard, K. Inhibition of cellular protein secretion by poliovirus proteins 2B and 3A. EMBO J. 1995, 14, 894–907. [Google Scholar] [CrossRef]
- Deitz, S.B.; Dodd, D.A.; Cooper, S.; Parham, P.; Kirkegaard, K. MHC I-dependent antigen presentation is inhibited by poliovirus protein 3A. Proc. Natl. Acad. Sci. USA 2000, 97, 13790–13795. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Langereis, M.A.; Van Kuppeveld, F.J. Induction and suppression of innate antiviral responses by picornaviruses. Cytokine Growth Factor Rev. 2014, 25, 577–585. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, J.; Li, Q. Immune Evasion of Enteroviruses Under Innate Immune Monitoring. Front. Microbiol. 2018, 9, 1866. [Google Scholar] [CrossRef]
- Flather, D.; Semler, B.L. Picornaviruses and nuclear functions: Targeting a cellular compartment distinct from the replication site of a positive-strand RNA virus. Front. Microbiol. 2015, 6, 594. [Google Scholar] [CrossRef] [Green Version]
- Gustin, K.E. Effects of poliovirus infection on nucleo-cytoplasmic trafficking and nuclear pore complex composition. EMBO J. 2001, 20, 240–249. [Google Scholar] [CrossRef] [Green Version]
- Gustin, K.E.; Sarnow, P. Inhibition of Nuclear Import and Alteration of Nuclear Pore Complex Composition by Rhinovirus. J. Virol. 2002, 76, 8787–8796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lidsky, P.V.; Hato, S.; Bardina, M.V.; Aminev, A.G.; Palmenberg, A.C.; Sheval, E.V.; Polyakov, V.Y.; Van Kuppeveld, F.J.M.; Agol, V.I. Nucleocytoplasmic Traffic Disorder Induced by Cardioviruses. J. Virol. 2006, 80, 2705–2717. [Google Scholar] [CrossRef] [Green Version]
- Viktorova, E.G.; Nchoutmboube, J.A.; Ford-Siltz, L.A.; Iverson, E.; Belov, G.A. Phospholipid synthesis fueled by lipid droplets drives the structural development of poliovirus replication organelles. PLoS Pathog. 2018, 14, e1007280. [Google Scholar] [CrossRef] [Green Version]
- Knowles, N.J.; Davies, P.R.; Henry, T.; O’Donnell, V.; Pacheco, J.M.; Mason, P.W. Emergence in Asia of Foot-and-Mouth Disease Viruses with Altered Host Range: Characterization of Alterations in the 3A Protein. J. Virol. 2001, 75, 1551–1556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, S.H.; Lee, K.-N.; Park, J.-H.; Kim, H. Molecular epidemiology of foot-and-mouth disease virus serotypes A and O with emphasis on Korean isolates: Temporal and spatial dynamics. Arch. Virol. 2011, 156, 817–826. [Google Scholar] [CrossRef]
- Greninger, A.L.; Knudsen, G.M.; Betegon, M.; Burlingame, A.L.; DeRisi, J.L. The 3A Protein from Multiple Picornaviruses Utilizes the Golgi Adaptor Protein ACBD3 To Recruit PI4KIII. J. Virol. 2012, 86, 3605–3616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, K.; Stoffel, W. TMbase—A database of membrane spanning proteins segments. Biol. Chem. Hoppe Seyler 1993, 374, 166. [Google Scholar]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden markov model: Application to complete genomes11Edited by F. Cohen. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Käll, L.; Krogh, A.; Sonnhammer, E.L. Advantages of combined transmembrane topology and signal peptide predictionߞThe Phobius web server. Nucleic Acids Res. 2007, 35, W429–W432. [Google Scholar] [CrossRef] [Green Version]
- Tusnady, G.E.; Simon, I. The HMMTOP transmembrane topology prediction server. Bioinformatics 2001, 17, 849–850. [Google Scholar] [CrossRef]
- Fujita, K.; Krishnakumar, S.S.; Franco, D.; Paul, A.V.; London, E.; Wimmer, E. Membrane Topography of the Hydrophobic Anchor Sequence of Poliovirus 3A and 3AB Proteins and the Functional Effect of 3A/3AB Membrane Association upon RNA Replication. Biochemestry 2007, 46, 5185–5199. [Google Scholar] [CrossRef] [Green Version]
- Wessels, E.; Duijsings, D.; Niu, T.-K.; Neumann, S.; Oorschot, V.M.; de Lange, F.; Lanke, K.H.; Klumperman, J.; Henke, A.; Jackson, C.L.; et al. A Viral Protein that Blocks Arf1-Mediated COP-I Assembly by Inhibiting the Guanine Nucleotide Exchange Factor GBF1. Dev. Cell 2006, 11, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Wessels, E.; Duijsings, D.; Lanke, K.H.W.; Melchers, W.J.G.; Jackson, C.L.; Van Kuppeveld, F.J.M. Molecular Determinants of the Interaction between Coxsackievirus Protein 3A and Guanine Nucleotide Exchange Factor GBF1. J. Virol. 2007, 81, 5238–5245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacheco, J.M.; Gladue, D.P.; Holinka, L.G.; Arzt, J.; Bishop, E.; Smoliga, G.; Pauszek, S.J.; Bracht, A.J.; O’Donnell, V.; Fernandez-Sainz, I.; et al. A partial deletion in non-structural protein 3A can attenuate foot-and-mouth disease virus in cattle. Virology 2013, 446, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Xiang, W.; Cuconati, A.; Paul, A.V.; Cao, X.; Wimmer, E. Molecular dissection of the multifunctional poliovirus RNA-binding protein 3AB. RNA 1995, 1, 892–904. [Google Scholar] [PubMed]
- Lawson, M.A.; Semler, B.L. Alternate poliovirus nonstructural protein processing cascades generated by primary sites of 3C proteinase cleavage. Virology 1992, 191, 309–320. [Google Scholar] [CrossRef]
- Pathak, H.B.; Oh, H.S.; Goodfellow, I.G.; Arnold, J.J.; Cameron, C.E. Picornavirus Genome Replication. J. Biol. Chem. 2008, 283, 30677–30688. [Google Scholar] [CrossRef] [Green Version]
- Leong, L.E.-C.; Cornell, C.T.; Semler, B.L. Processing Determinants and Functions of Cleavage Products of Picornavirus Polyproteins. In Molecular Biology of Picornavirus; American Society for Microbiology Press: Washington, DC, USA, 2014; pp. 185–197. [Google Scholar]
- O’Donnell, V.K.; Pacheco, J.M.; Henry, T.M.; Mason, P.W. Subcellular Distribution of the Foot-and-Mouth Disease Virus 3A Protein in Cells Infected with Viruses Encoding Wild-Type and Bovine-Attenuated Forms of 3A. Virology 2001, 287, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.V. Possible Unifying Mechanism of Picornavirus Genome Replication. In Molecular Biology of Picornavirus; American Society for Microbiology Press: Washington, DC, USA, 2014; pp. 225–246. [Google Scholar]
- Strauss, D.M.; Glustrom, L.W.; Wuttke, D.S. Towards an Understanding of the Poliovirus Replication Complex: The Solution Structure of the Soluble Domain of the Poliovirus 3A Protein. J. Mol. Biol. 2003, 330, 225–234. [Google Scholar] [CrossRef]
- González-Magaldi, M.; Postigo, R.; De La Torre, B.G.; Vieira, Y.A.; Rodríguez-Pulido, M.; López-Viñas, E.; Gómez-Puertas, P.; Andreu, D.; Kremer, L.; Rosas, M.F.; et al. Mutations That Hamper Dimerization of Foot-and-Mouth Disease Virus 3A Protein Are Detrimental for Infectivity. J. Virol. 2012, 86, 11013–11023. [Google Scholar] [CrossRef] [Green Version]
- Wessels, E.; Notebaart, R.A.; Duijsings, D.; Lanke, K.; Vergeer, B.; Melchers, W.J.G.; van Kuppeveld, F.J.M. Structure-Function Analysis of the Coxsackievirus Protein 3A. J. Biol. Chem. 2006, 281, 28232–28243. [Google Scholar] [CrossRef] [Green Version]
- Horova, V.; Lyoo, H.; Różycki, B.; Chalupska, D.; Smola, M.; Humpolickova, J.; Strating, J.R.P.M.; Van Kuppeveld, F.J.M.; Boura, E.; Klima, M. Convergent evolution in the mechanisms of ACBD3 recruitment to picornavirus replication sites. PLoS Pathog. 2019, 15, e1007962. [Google Scholar] [CrossRef] [Green Version]
- Hope, D.A.; Diamond, S.E.; Kirkegaard, K. Genetic dissection of interaction between poliovirus 3D polymerase and viral protein 3AB. J. Virol. 1997, 71, 9490–9498. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Sasaki, J.; Taniguchi, K. Overall linkage map of the nonstructural proteins of Aichi virus. Virus Res. 2010, 147, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Teterina, N.L.; Levenson, E.; Rinaudo, M.S.; Egger, D.; Bienz, K.; Gorbalenya, A.E.; Ehrenfeld, E. Evidence for Functional Protein Interactions Required for Poliovirus RNA Replication. J. Virol. 2006, 80, 5327–5337. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, W.; Cuconati, A.; Hope, D.; Kirkegaard, K.; Wimmer, E. Complete Protein Linkage Map of Poliovirus P3 Proteins: Interaction of Polymerase 3Dpol with VPg and with Genetic Variants of 3AB. J. Virol. 1998, 72, 6732–6741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, J.; Liu, Y.; Wimmer, E.; Paul, A.V. Complete protein linkage map between the P2 and P3 non-structural proteins of poliovirus. J. Gen. Virol. 2007, 88, 2259–2267. [Google Scholar] [CrossRef] [PubMed]
- Teterina, N.L.; Pinto, Y.; Weaver, J.D.; Jensen, K.S.; Ehrenfeld, E. Analysis of Poliovirus Protein 3A Interactions with Viral and Cellular Proteins in Infected Cells. J. Virol. 2011, 85, 4284–4296. [Google Scholar] [CrossRef] [Green Version]
- Towner, J.S.; Brown, D.M.; Nguyen, J.H.; Semler, B.L. Functional conservation of the hydrophobic domain of polypeptide 3AB between human rhinovirus and poliovirus. Virol. 2003, 314, 432–442. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, Y.; Shan, C.; Sun, Y.; Xu, P.; Zhou, H.; Yang, C.; Shi, P.-Y.; Rao, Z.; Zhang, B.; et al. Crystal Structure of Enterovirus 71 RNA-Dependent RNA Polymerase Complexed with Its Protein Primer VPg: Implication for a trans Mechanism of VPg Uridylylation. J. Virol. 2013, 87, 5755–5768. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Orta, C.; Arias, A.; Agudo, R.; Pérez-Luque, R.; Escarmís, C.; Domingo, E.; Verdaguer, N. The structure of a protein primer–polymerase complex in the initiation of genome replication. EMBO J. 2006, 25, 880–888. [Google Scholar] [CrossRef]
- Gruez, A.; Selisko, B.; Roberts, M.; Bricogne, G.; Bussetta, C.; Jabafi, I.; Coutard, B.; De Palma, A.M.; Neyts, J.; Canard, B. The Crystal Structure of Coxsackievirus B3 RNA-Dependent RNA Polymerase in Complex with Its Protein Primer VPg Confirms the Existence of a Second VPg Binding Site on Picornaviridae Polymerases. J. Virol. 2008, 82, 9577–9590. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Orta, C.; Ferrero, D.; Verdaguer, N. RNA-Dependent RNA Polymerases of Picornaviruses: From the Structure to Regulatory Mechanisms. Viruses 2015, 7, 4438–4460. [Google Scholar] [CrossRef] [Green Version]
- Molla, A.; Harris, K.S.; Paul, A.V.; Shin, S.H.; Mugavero, J.; Wimmer, E. Stimulation of poliovirus proteinase 3Cpro-related proteolysis by the genome-linked protein VPg and its precursor 3AB. J. Biol. Chem. 1994, 269, 27015–27020. [Google Scholar] [CrossRef]
- Nagashima, S.; Sasaki, J.; Taniguchi, K. Interaction between Polypeptide 3ABC and the 5′-Terminal Structural Elements of the Genome of Aichi Virus: Implication for Negative-Strand RNA Synthesis. J. Virol. 2008, 82, 6161–6171. [Google Scholar] [CrossRef] [Green Version]
- Moffat, K.; Howell, G.; Knox, C.; Belsham, G.J.; Monaghan, P.; Ryan, M.D.; Wileman, T. Effects of Foot-and-Mouth Disease Virus Nonstructural Proteins on the Structure and Function of the Early Secretory Pathway: 2BC but Not 3A Blocks Endoplasmic Reticulum-to-Golgi Transport. J. Virol. 2005, 79, 4382–4395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wessels, E.; Duijsings, D.; Notebaart, R.A.; Melchers, W.J.G.; Van Kuppeveld, F.J.M. A Proline-Rich Region in the Coxsackievirus 3A Protein Is Required for the Protein To Inhibit Endoplasmic Reticulum-to-Golgi Transport. J. Virol. 2005, 79, 5163–5173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dodd, D.A.; Giddings, T.H., Jr.; Kirkegaard, K. Poliovirus 3A Protein Limits Interleukin-6 (IL-6), IL-8, and Beta Interferon Secretion during Viral Infection. J. Virol. 2001, 75, 8158–8165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doedens, J.R.; Giddings, T.H.; Kirkegaard, K. Inhibition of endoplasmic reticulum-to-Golgi traffic by poliovirus protein 3A: Genetic and ultrastructural analysis. J. Virol. 1997, 71, 9054–9064. [Google Scholar] [CrossRef] [Green Version]
- Neznanov, N.; Kondratova, A.; Chumakov, K.M.; Angres, B.; Zhumabayeva, B.; Agol, V.I.; Gudkov, A.V. Poliovirus Protein 3A Inhibits Tumor Necrosis Factor (TNF)-Induced Apoptosis by Eliminating the TNF Receptor from the Cell Surface. J. Virol. 2001, 75, 10409–10420. [Google Scholar] [CrossRef] [Green Version]
- Moffat, K.; Knox, C.; Howell, G.; Clark, S.J.; Yang, H.; Belsham, G.J.; Ryan, M.D.; Wileman, T. Inhibition of the Secretory Pathway by Foot-and-Mouth Disease Virus 2BC Protein Is Reproduced by Coexpression of 2B with 2C, and the Site of Inhibition Is Determined by the Subcellular Location of 2C. J. Virol. 2006, 81, 1129–1139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choe, S.S.; Dodd, D.A.; Kirkegaard, K. Inhibition of cellular protein secretion by picornaviral 3A proteins. Virology 2005, 337, 18–29. [Google Scholar] [CrossRef] [Green Version]
- Wessels, E.; Duijsings, D.; Lanke, K.H.W.; Van Dooren, S.H.J.; Jackson, C.L.; Melchers, W.J.G.; Van Kuppeveld, F.J.M. Effects of Picornavirus 3A Proteins on Protein Transport and GBF1-Dependent COP-I Recruitment. J. Virol. 2006, 80, 11852–11860. [Google Scholar] [CrossRef] [Green Version]
- Mousnier, A.; Swieboda, D.; Pinto, A.; Guedán, A.; Rogers, A.V.; Walton, R.; Johnston, S.L.; Solari, R. Human Rhinovirus 16 Causes Golgi Apparatus Fragmentation without Blocking Protein Secretion. J. Virol. 2014, 88, 11671–11685. [Google Scholar] [CrossRef] [Green Version]
- Beske, O.; Reichelt, M.; Taylor, M.P.; Kirkegaard, K.; Andino, R. Poliovirus infection blocks ERGIC-to-Golgi trafficking and induces microtubule-dependent disruption of the Golgi complex. J. Cell Sci. 2007, 120, 3207–3218. [Google Scholar] [CrossRef] [Green Version]
- Szul, T.; Sztul, E. COPII and COPI Traffic at the ER-Golgi Interface. Physiol. 2011, 26, 348–364. [Google Scholar] [CrossRef] [PubMed]
- Szul, T.; Garcia-Mata, R.; Brandon, E.; Shestopal, S.; Alvarez, C.; Sztul, E. Dissection of Membrane Dynamics of the ARF-Guanine Nucleotide Exchange Factor GBF1. Traffic 2005, 6, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Sztul, E.; Chen, P.-W.; Casanova, J.E.; Cherfils, J.; Dacks, J.B.; Lambright, D.G.; Lee, F.-J.S.; Randazzo, P.A.; Santy, L.C.; Schürmann, A.; et al. ARF GTPases and their GEFs and GAPs: Concepts and challenges. Mol. Biol. Cell 2019, 30, 1249–1271. [Google Scholar] [CrossRef] [PubMed]
- Niu, T.-K.; Pfeifer, A.C.; Lippincott-Schwartz, J.; Jackson, C.L. Dynamics of GBF1, a Brefeldin A-Sensitive Arf1 Exchange Factor at the Golgi. Mol. Biol. Cell 2005, 16, 1213–1222. [Google Scholar] [CrossRef] [Green Version]
- Arita, M.; Wakita, T.; Shimizu, H. Valosin-Containing Protein (VCP/p97) Is Required for Poliovirus Replication and Is Involved in Cellular Protein Secretion Pathway in Poliovirus Infection. J. Virol. 2012, 86, 5541–5553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berstein, H.D.; Baltimore, D. Poliovirus mutant that contains a cold-sensitive defect in viral RNA synthesis. J. Virol. 1988, 62, 2922–2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, U.; Dasgupta, A. Expression and subcellular localization of poliovirus VPg-precursor protein 3AB in eukaryotic cells: Evidence for glycosylation in vitro. J. Virol. 1994, 68, 4468–4477. [Google Scholar] [CrossRef] [Green Version]
- Giachetti, C.; Hwang, S.S.; Semler, B.L. cis-acting lesions targeted to the hydrophobic domain of a poliovirus membrane protein involved in RNA replication. J. Virol. 1992, 66, 6045–6057. [Google Scholar] [CrossRef] [Green Version]
- Semler, B.L.; Anderson, C.W.; Hanecak, R.; Dorner, L.F.; Wimmer, E. A membrane-associated precursor to poliovirus VPg identified by immunoprecipitation with antibodies directed against a synthetic heptapeptide. Cell 1982, 28, 405–412. [Google Scholar] [CrossRef]
- Towner, J.S.; Ho, T.V.; Semler, B.L. Determinants of Membrane Association for Poliovirus Protein 3AB. J. Biol. Chem. 1996, 271, 26810–26818. [Google Scholar] [CrossRef] [Green Version]
- Towner, J.S.; Mazanet, M.M.; Semler, B.L. Rescue of Defective Poliovirus RNA Replication by 3AB-Containing Precursor Polyproteins. J. Virol. 1998, 72, 7191–7200. [Google Scholar] [CrossRef] [Green Version]
- González-Magaldi, M.; Martín-Acebes, M.A.; Kremer, L.; Sobrino, F. Membrane Topology and Cellular Dynamics of Foot-and-Mouth Disease Virus 3A Protein. PLoS ONE 2014, 9, e106685. [Google Scholar] [CrossRef]
- Lotufo, C.M.; Wilda, M.; Giraldez, A.N.; Grigera, P.R.; Mattion, N.M. Relevance of the N-terminal and major hydrophobic domains of non-structural protein 3A in the replicative process of a DNA-launched foot-and-mouth disease virus replicon. Arch. Virol. 2018, 163, 1769–1778. [Google Scholar] [CrossRef]
- Racaniello, V.R. One hundred years of poliovirus pathogenesis. Virology 2006, 344, 9–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arzt, J.; Baxt, B.; Grubman, M.J.; Jackson, T.; Juleff, N.; Rhyan, J.; Rieder, E.; Waters, R.; Rodriguez, L.L. The Pathogenesis of Foot-and-Mouth Disease II: Viral Pathways in Swine, Small Ruminants, and Wildlife; Myotropism, Chronic Syndromes, and Molecular Virus-Host Interactions. Transbound. Emerg. Dis. 2011, 58, 305–326. [Google Scholar] [CrossRef]
- Weaver, G.V.; Domenech, J.; Thiermann, A.R.; Karesh, W.B. Foot and Mouth Disease: A Look from the Wild Side. J. Wildl. Dis. 2013, 49, 759–785. [Google Scholar] [CrossRef]
- Harris, J.R.; Racaniello, V.R. Amino Acid Changes in Proteins 2B and 3A Mediate Rhinovirus Type 39 Growth in Mouse Cells. J. Virol. 2005, 79, 5363–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen, A.L.; Racaniello, V.R. Selection of rhinovirus 1A variants adapted for growth in mouse lung epithelial cells. Virology 2011, 420, 82–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bochkov, Y.A.; Watters, K.; Basnet, S.; Sijapati, S.; Hill, M.; Palmenberg, A.C.; Gern, J.E. Mutations in VP1 and 3A proteins improve binding and replication of rhinovirus C15 in HeLa-E8 cells. Virology 2016, 499, 350–360. [Google Scholar] [CrossRef]
- Graff, J.; Kasang, C.; Normann, A.; Pfisterer-Hunt, M.; Feinstone, S.M.; Flehmig, B. Mutational Events in Consecutive Passages of Hepatitis A Virus Strain GBM during Cell Culture Adaptation. Virology 1994, 204, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Normann, A.; Feinstone, S.M.; Flehmig, B. Nucleotide sequence of wild-type hepatitis A virus GBM in comparison with two cell culture-adapted variants. J. Virol. 1994, 68, 548–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemon, S.M.; Murphy, P.C.; Shields, P.A.; Ping, L.H.; Feinstone, S.M.; Cromeans, T.; Jansen, R.W. Antigenic and genetic variation in cytopathic hepatitis A virus variants arising during persistent infection: Evidence for genetic recombination. J. Virol. 1991, 65, 2056–2065. [Google Scholar] [CrossRef] [Green Version]
- Morace, G.; Pisani, G.; Beneduce, F.; Divizia, M.; Pana, A. Mutations in the 3A genomic region of two cytopathic strains of hepatitis A virus isolated in Italy. Virus Res. 1993, 28, 187–194. [Google Scholar] [CrossRef]
- Massilamany, C.; Gangaplara, A.; Basavalingappa, R.H.; Rajasekaran, R.A.; Vu, H.; Riethoven, J.-J.; Steffen, D.; Pattnaik, A.K.; Reddy, J. Mutations in the 5’ NTR and the Non-Structural Protein 3A of the Coxsackievirus B3 Selectively Attenuate Myocarditogenicity. PLoS ONE 2015, 10, e0131052. [Google Scholar] [CrossRef] [Green Version]
- Núñez, J.I.; Baranowski, E.; Molina, N.; Ruiz-Jarabo, C.M.; Sánchez, C.; Domingo, E.; Sobrino, F. A Single Amino Acid Substitution in Nonstructural Protein 3A Can Mediate Adaptation of Foot-and-Mouth Disease Virus to the Guinea Pig. J. Virol. 2001, 75, 3977–3983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giraudo, A.T.; Beck, E.; Strebel, K.; De Mello, P.A.; La Torre, J.; Scodeller, E.A.; Bergmann, I.E. Identification of a nucleotide deletion in parts of polypeptide 3A in two independent attenuated aphthovirus strains. Virology 1990, 177, 780–783. [Google Scholar] [CrossRef]
- Giraudo, A.T.; Sagedahl, A.; Bergmann, I.E.; La Torre, J.L.; Scodeller, E.A. Isolation and characterization of recombinants between attenuated and virulent aphthovirus strains. J. Virol. 1987, 61, 419–425. [Google Scholar] [CrossRef] [Green Version]
- Parisi, J.M.; Giomi, P.C.; Grigera, P.; Mello, P.A.; Bergmann, I.E.; La Torre, J.L.; Scodeller, E.A. Biochemical characterization of an aphthovirus type 01 strain campos attenuated for cattle by serial passages in chicken embryos. Virology 1985, 147, 61–71. [Google Scholar] [CrossRef]
- Sagedahl, A.; Giraudo, A.T.; De Mellod, P.A.; Bergmann, I.E.; La Torre, J.; Scodeller, E.A. Biochemical characterization of an aphthovirus type C3 strain resende attenuated for cattle by serial passages in chicken embryos. Virology 1987, 157, 366–374. [Google Scholar] [CrossRef]
- Beard, C.W.; Mason, P.W. Genetic Determinants of Altered Virulence of Taiwanese Foot-and-Mouth Disease Virus. J. Virol. 2000, 74, 987–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, C.S.; Donaldson, A.I. Natural adaption to pigs of a Taiwanese isolate of foot-and-mouth disease virus. Veter Rec. 1997, 141, 174–175. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Gao, M.; Zhang, R.; Song, G.; Song, J.; Liu, D.; Cao, Y.; Li, T.; Ma, B.; Liu, X.; et al. A mutant of infectious Asia 1 serotype foot-and-mouth disease virus with the deletion of 10-amino-acid in the 3A protein. Virus Genes 2010, 41, 406–413. [Google Scholar] [CrossRef]
- Pacheco, J.M.; Henry, T.M.; O’Donnell, V.K.; Gregory, J.B.; Mason, P.W. Role of Nonstructural Proteins 3A and 3B in Host Rangeand Pathogenicity of Foot-and-Mouth Disease Virus. J. Virol. 2003, 77, 13017–13027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larocco, M.; Krug, P.W.; Kramer, E.; Ahmed, Z.; Pacheco, J.M.; Duque, H.; Baxt, B.; Rodriguez, L.L. Correction for LaRocco et al., A Continuous Bovine Kidney Cell Line Constitutively Expressing Bovine αVβ6Integrin Has Increased Susceptibility to Foot-and-Mouth Disease Virus. J. Clin. Microbiol. 2015, 53, 755. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Li, P.; Bai, X.; Sun, P.; Bao, H.; Lu, Z.; Cao, Y.; Li, D.; Chen, Y.; Qiao, Z.; et al. Sequences outside that of residues 93–102 of 3A protein can contribute to the ability of foot-and-mouth disease virus (FMDV) to replicate in bovine-derived cells. Virus Res. 2014, 191, 161–171. [Google Scholar] [CrossRef]
- Stenfeldt, C.; Arzt, J.; Pacheco, J.M.; Gladue, D.P.; Smoliga, G.R.; Silva, E.B.; Rodriguez, L.L.; Borca, M.V. A partial deletion within foot-and-mouth disease virus non-structural protein 3A causes clinical attenuation in cattle but does not prevent subclinical infection. Virology 2018, 516, 115–126. [Google Scholar] [CrossRef]
- DeStefano, J.J.; Titilope, O. Poliovirus Protein 3AB Displays Nucleic Acid Chaperone and Helix-Destabilizing Activities. J. Virol. 2006, 80, 1662–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gangaramani, D.R.; Eden, E.L.; Shah, M.; DeStefano, J.J. The twenty-nine amino acid C-terminal cytoplasmic domain of poliovirus 3AB is critical for nucleic acid chaperone activity. RNA Biol. 2010, 7, 820–829. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Xia, H.; Wang, P.; Yang, J.; Zhao, T.; Zhang, Q.; Hu, Y.; Zhou, X. The identification and characterization of nucleic acid chaperone activity of human enterovirus 71 nonstructural protein 3AB. Virology 2014, 464–465, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Kondratova, A.A.; Neznanov, N.; Kondratov, R.V.; Gudkov, A.V. Poliovirus Protein 3A Binds and Deregulates LIS1, Causing Block of Membrane Protein Trafficking and Deregulation of Cell Division. Cell Cycle 2005, 4, 1403–1410. [Google Scholar] [CrossRef] [Green Version]
- Htet, Z.M.; Gillies, J.P.; Baker, R.W.; Leschziner, A.E.; DeSantis, M.E.; Reck-Peterson, S.L. LIS1 promotes the formation of activated cytoplasmic dynein-1 complexes. Nat. Cell Biol. 2020, 22, 518–525. [Google Scholar] [CrossRef]
- Toropova, K.; Zou, S.; Roberts, A.J.; Redwine, W.B.; Goodman, B.S.; Reck-Peterson, S.L.; E Leschziner, A. Lis1 regulates dynein by sterically blocking its mechanochemical cycle. eLife 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.J.; Tan, K.; Reck-Peterson, S.L. Lis1 is an initiation factor for dynein-driven organelle transport. J. Cell Biol. 2012, 197, 971–982. [Google Scholar] [CrossRef]
- Reck-Peterson, S.L.; Redwine, W.B.; Vale, R.D.; Carter, A.P. The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol. 2018, 19, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Gladue, D.P.; O’Donnell, V.; Baker-Bransetter, R.; Pacheco, J.M.; Holinka, L.G.; Arzt, J.; Pauszek, S.; Fernandez-Sainz, I.; Fletcher, P.; Brocchi, E.; et al. Interaction of Foot-and-Mouth Disease Virus Nonstructural Protein 3A with Host Protein DCTN3 Is Important for Viral Virulence in Cattle. J. Virol. 2013, 88, 2737–2747. [Google Scholar] [CrossRef] [Green Version]
- Santos, T.D.L.; Botton, S.D.A.; Weiblen, R.; Grubman, M.J. The Leader Proteinase of Foot-and-Mouth Disease Virus Inhibits the Induction of Beta Interferon mRNA and Blocks the Host Innate Immune Response. J. Virol. 2006, 80, 1906–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowery, J.; Kuczmarski, E.R.; Herrmann, H.; Goldman, R.D. Intermediate Filaments Play a Pivotal Role in Regulating Cell Architecture and Function. J. Biol. Chem. 2015, 290, 17145–17153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wen, Z.; Shi, X.; Liu, Y.-J.; Eriksson, J.E.; Jiu, Y. The diverse roles and dynamic rearrangement of vimentin during viral infection. J. Cell Sci. 2021, 134, jcs250597. [Google Scholar] [CrossRef]
- Ma, X.; Ling, Y.; Li, P.; Sun, P.; Cao, Y.; Bai, X.; Li, K.; Fu, Y.; Zhang, J.; Li, D.; et al. Cellular Vimentin Interacts with Foot-and-Mouth Disease Virus Nonstructural Protein 3A and Negatively Modulates Viral Replication. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.; Barnett, P.V.; Denyer, M.S.; Jackson, T.; Stirling, C.M.A.; Hawes, P.C.; Simpson, J.L.; Monaghan, P.; Takamatsu, H.-H. Foot-and-Mouth Disease Virus Replicates Only Transiently in Well-Differentiated Porcine Nasal Epithelial Cells. J. Virol. 2010, 84, 9149–9160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilles, C.; Polette, M.; Zahm, J.M.; Tournier, J.M.; Volders, L.; Foidart, J.M.; Birembaut, P. Vimentin contributes to human mammary epithelial cell migration. J. Cell Sci. 1999, 112, 4615–4625. [Google Scholar] [PubMed]
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef]
- Hur, S. Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu. Rev. Immunol. 2019, 37, 349–375. [Google Scholar] [CrossRef]
- Barral, P.M.; Morrison, J.M.; Drahos, J.; Gupta, P.; Sarkar, D.; Fisher, P.B.; Racaniello, V.R. MDA-5 Is Cleaved in Poliovirus-Infected Cells. J. Virol. 2007, 81, 3677–3684. [Google Scholar] [CrossRef] [Green Version]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro Targets MDA5 and MAVS in Infected Cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef] [Green Version]
- Pulido, M.R.; Martínez-Salas, E.; Sobrino, F.; Sáiz, M. MDA5 cleavage by the Leader protease of foot-and-mouth disease virus reveals its pleiotropic effect against the host antiviral response. Cell Death Dis. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, L.; Stipkovits, L.; Szathmary, S.; Li, X.; Liu, Y. The Strategy of Picornavirus Evading Host Antiviral Responses: Non-structural Proteins Suppress the Production of IFNs. Front. Microbiol. 2018, 9, 2943. [Google Scholar] [CrossRef]
- Kumar, H.; Kawai, T.; Kato, H.; Sato, S.; Takahashi, K.; Coban, C.; Yamamoto, M.; Uematsu, S.; Ishii, K.J.; Takeuchi, O.; et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J. Exp. Med. 2006, 203, 1795–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malik, N.; Canfield, V.A.; Beckers, M.-C.; Gros, P.; Levenson, R. Identification of the Mammalian Na,K-ATPase β3 Subunit. J. Biol. Chem. 1996, 271, 22754–22758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Y.; Hou, H.; Wang, F.; Qiao, L.; Wang, X.; Yu, J.; Liu, W.; Sun, Z. ATP1B3: A virus-induced host factor against EV71 replication by up-regulating the production of type-I interferons. Virology 2016, 496, 28–34. [Google Scholar] [CrossRef]
- Zheng, B.; Zhang, J.; Zheng, T.; Wang, H.; Li, Z.; Huan, C.; Ning, S.; Wang, Y.; Zhang, W. ATP1B3 cooperates with BST-2 to promote hepatitis B virus restriction. J. Med Virol. 2020, 92, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Lei, C.; Xu, Z.; Yang, F.; Liu, H.; Zhu, Z.; Li, S.; Liu, X.; Shu, H.; Zheng, H. Foot-and-mouth disease virus non-structural protein 3A inhibits the interferon-β signaling pathway. Sci. Rep. 2016, 6, 21888. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Bi, J.; Liu, J.; Liu, X.; Wu, X.; Jiang, P.; Yoo, D.; Zhang, Y.; Wu, J.; Wan, R.; et al. 3Cpro of Foot-and-Mouth Disease Virus Antagonizes the Interferon Signaling Pathway by Blocking STAT1/STAT2 Nuclear Translocation. J. Virol. 2014, 88, 4908–4920. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Fu, S.; Wu, Z.; Yang, W.; Ru, Y.; Shu, H.; Liu, X.; Zheng, H. DDX56 inhibits type I interferon by disrupting assembly of IRF3–IPO5 to inhibit IRF3 nucleus import. J. Cell Sci. 2020, 133, jcs230409. [Google Scholar] [CrossRef] [Green Version]
- Fu, S.-Z.; Yang, W.-P.; Ru, Y.; Zhang, K.-S.; Wang, Y.; Liu, X.-T.; Li, D.; Zheng, H.-X. DDX56 cooperates with FMDV 3A to enhance FMDV replication by inhibiting the phosphorylation of IRF3. Cell. Signal. 2019, 64, 109393. [Google Scholar] [CrossRef]
- Reineke, L.C.; Kedersha, N.; Langereis, M.A.; Van Kuppeveld, F.J.M.; Lloyd, R.E. Stress Granules Regulate Double-Stranded RNA-Dependent Protein Kinase Activation through a Complex Containing G3BP1 and Caprin1. mBio 2015, 6, e02486. [Google Scholar] [CrossRef] [Green Version]
- Reineke, L.C.; Lloyd, R.E. The Stress Granule Protein G3BP1 Recruits Protein Kinase R To Promote Multiple Innate Immune Antiviral Responses. J. Virol. 2015, 89, 2575–2589. [Google Scholar] [CrossRef] [Green Version]
- Onomoto, K.; Yoneyama, M.; Fung, G.; Kato, H.; Fujita, T. Antiviral innate immunity and stress granule responses. Trends Immunol. 2014, 35, 420–428. [Google Scholar] [CrossRef] [PubMed]
- Eiermann, N.; Haneke, K.; Sun, Z.; Stoecklin, G.; Ruggieri, A. Dance with the Devil: Stress Granules and Signaling in Antiviral Responses. Viruses 2020, 12, 984. [Google Scholar] [CrossRef]
- Kim, S.S.-Y.; Sze, L.; Lam, K.-P. The stress granule protein G3BP1 binds viral dsRNA and RIG-I to enhance interferon-β response. J. Biol. Chem. 2019, 294, 6430–6438. [Google Scholar] [CrossRef] [Green Version]
- Yang, W.; Li, D.; Ru, Y.; Bai, J.; Ren, J.; Zhang, J.; Li, L.; Liu, X.; Zheng, H. Foot-and-Mouth Disease Virus 3A Protein Causes Upregulation of Autophagy-Related Protein LRRC25 To Inhibit the G3BP1-Mediated RIG-Like Helicase-Signaling Pathway. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [Green Version]
- Galan, A.; Lozano, G.; Piñeiro, D.; Martinez-Salas, E. G3BP1 interacts directly with the FMDV IRES and negatively regulates translation. FEBS J. 2017, 284, 3202–3217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, X.; Pan, T.; Wang, D.; Fang, L.; Ma, J.; Zhu, X.; Shi, Y.; Zhang, K.; Zheng, H.; Chen, H.; et al. Foot-and-Mouth Disease Virus Counteracts on Internal Ribosome Entry Site Suppression by G3BP1 and Inhibits G3BP1-Mediated Stress Granule Assembly via Post-Translational Mechanisms. Front. Immunol. 2018, 9, 1142. [Google Scholar] [CrossRef] [Green Version]
- Visser, L.J.; Medina, G.N.; Rabouw, H.H.; De Groot, R.J.; Langereis, M.A.; Santos, T.D.L.; Van Kuppeveld, F.J.M. Foot-and-Mouth Disease Virus Leader Protease Cleaves G3BP1 and G3BP2 and Inhibits Stress Granule Formation. J. Virol. 2018, 93, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maillard, P.V.; Van Der Veen, A.G.; Poirier, E.Z.; Sousa, C.R.E. Slicing and dicing viruses: Antiviral RNA interference in mammals. EMBO J. 2019, 38. [Google Scholar] [CrossRef]
- Qiu, Y.; Xu, Y.; Zhang, Y.; Zhou, H.; Deng, Y.-Q.; Li, X.-F.; Miao, M.; Zhang, Q.; Zhong, B.; Hu, Y.; et al. Human Virus-Derived Small RNAs Can Confer Antiviral Immunity in Mammals. Immunology 2017, 46, 992–1004. [Google Scholar] [CrossRef] [Green Version]
- Mu, J.; Zhang, H.; Li, T.; Shu, T.; Qiu, Y.; Zhou, X. The 3A protein of coxsackievirus B3 acts as a viral suppressor of RNA interference. J. Gen. Virol. 2020, 101, 1069–1078. [Google Scholar] [CrossRef]
- Schuster, S.; Overheul, G.J.; Bauer, L.; Van Kuppeveld, F.J.M.; Van Rij, R.P. No evidence for viral small RNA production and antiviral function of Argonaute 2 in human cells. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gamarnik, A.V.; Andino, R. Switch from translation to RNA replication in a positive-stranded RNA virus. Genes Dev. 1998, 12, 2293–2304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Novak, J.E.; Kirkegaard, K. Coupling between genome translation and replication in an RNA virus. Genes Dev. 1994, 8, 1726–1737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, K.S.; Xiang, W.; Alexander, L.; Lane, W.S.; Paul, A.V.; Wimmer, E. Interaction of poliovirus polypeptide 3CDpro with the 5‘ and 3‘ termini of the poliovirus genome. Identification of viral and cellular cofactors needed for efficient binding. J. Biol. Chem. 1994, 269, 27004–27014. [Google Scholar] [CrossRef]
- Parsley, T.B.; Towner, J.S.; Blyn, L.B.; Ehrenfeld, E.; Semler, B.L. Poly (rC) binding protein 2 forms a ternary complex with the 5’-terminal sequences of poliovirus RNA and the viral 3CD proteinase. RNA 1997, 3, 1124–1134. [Google Scholar] [PubMed]
- Xiang, W.; Harris, K.S.; Alexander, L.; Wimmer, E. Interaction between the 5’-terminal cloverleaf and 3AB/3CDpro of poliovirus is essential for RNA replication. J. Virol. 1995, 69, 3658–3667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, A.V.; Cao, X.; Harris, K.S.; Lama, J.; Wimmer, E. Studies with poliovirus polymerase 3Dpol. Stimulation of poly(U) synthesis in vitro by purified poliovirus protein 3AB. J. Biol. Chem. 1994, 269, 29173–29181. [Google Scholar] [CrossRef]
- Spear, A.R.; Ogram, S.A.; Morasco, B.J.; Smerage, L.E.; Flanegan, J.B. Viral precursor protein P3 and its processed products perform discrete and essential functions in the poliovirus RNA replication complex. Virology 2015, 485, 492–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Franco, D.; Paul, A.V.; Wimmer, E. Tyrosine 3 of Poliovirus Terminal Peptide VPg(3B) Has an Essential Function in RNA Replication in the Context of Its Precursor Protein, 3AB. J. Virol. 2007, 81, 5669–5684. [Google Scholar] [CrossRef] [Green Version]
- Herod, M.R.; Loundras, E.-A.; Ward, J.C.; Tulloch, F.; Rowlands, D.J.; Stonehouse, N.J. Employing transposon mutagenesis to investigate foot-and-mouth disease virus replication. J. Gen. Virol. 2015, 96, 3507–3518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teterina, N.L.; Rinaudo, M.S.; Ehrenfeld, E. Strand-Specific RNA Synthesis Defects in a Poliovirus with a Mutation in Protein 3A. J. Virol. 2003, 77, 12679–12691. [Google Scholar] [CrossRef] [Green Version]
- Lyle, J.M.; Clewell, A.; Richmond, K.; Richards, O.C.; Hope, D.A.; Schultz, S.C.; Kirkegaard, K. Similar Structural Basis for Membrane Localization and Protein Priming by an RNA-dependent RNA Polymerase. J. Biol. Chem. 2002, 277, 16324–16331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lama, J.; Paul, A.; Harris, K.; Wimmer, E. Properties of purified recombinant poliovirus protein 3aB as substrate for viral proteinases and as co-factor for RNA polymerase 3Dpol. J. Biol. Chem. 1994, 269, 66–70. [Google Scholar] [CrossRef]
- Plotch, S.J.; Palant, O. Poliovirus protein 3AB forms a complex with and stimulates the activity of the viral RNA polymerase, 3Dpol. J. Virol. 1995, 69, 7169–7179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richards, O.C.; Ehrenfeld, E. Effects of Poliovirus 3AB Protein on 3D Polymerase-catalyzed Reaction. J. Biol. Chem. 1998, 273, 12832–12840. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez-Wells, V. Primer-dependent synthesis by poliovirus RNA-dependent RNA polymerase (3Dpol). Nucleic Acids Res. 2001, 29, 2715–2724. [Google Scholar] [CrossRef] [Green Version]
- Lama, J.; Sanz, M.A.; Rodrguez, P.L. A Role for 3AB Protein in Poliovirus Genome Replication. J. Biol. Chem. 1995, 270, 14430–14438. [Google Scholar] [CrossRef] [Green Version]
- Strauss, D.M.; Wuttke, D.S. Characterization of Protein-Protein Interactions Critical for Poliovirus Replication: Analysis of 3AB and VPg Binding to the RNA-Dependent RNA Polymerase. J. Virol. 2007, 81, 6369–6378. [Google Scholar] [CrossRef] [Green Version]
- Takegami, T.; Kuhn, R.J.; Anderson, C.W.; Wimmer, E. Membrane-dependent uridylylation of the genome-linked protein VPg of poliovirus. Proc. Natl. Acad. Sci. USA 1983, 80, 7447–7451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takegami, T.; Semler, B.L.; Anderson, C.W.; Wimmer, E. Membrane fractions active in poliovirus RNA replication contain VPg precursor polypeptides. Virol. 1983, 128, 33–47. [Google Scholar] [CrossRef]
- Hall, D.J.; Palmenberg, A.C. Cleavage site mutations in the encephalomyocarditis virus P3 region lethally abrogate the normal processing cascade. J. Virol. 1996, 70, 5954–5961. [Google Scholar] [CrossRef] [Green Version]
- Nayak, A.; Goodfellow, I.G.; Woolaway, K.E.; Birtley, J.; Curry, S.; Belsham, G.J. Role of RNA Structure and RNA Binding Activity of Foot-and-Mouth Disease Virus 3C Protein in VPg Uridylylation and Virus Replication. J. Virol. 2006, 80, 9865–9875. [Google Scholar] [CrossRef] [Green Version]
- Bienz, K.; Egger, D.; Pasamontes, L. Association of polioviral proteins of the P2 genomic region with the viral replication complex and virus-induced membrane synthesis as visualized by electron microscopic immunocytochemistry and autoradiography. Virology 1987, 160, 220–226. [Google Scholar] [CrossRef]
- Dorobantu, C.M.; Albulescu, L.; Harak, C.; Feng, Q.; Van Kampen, M.; Strating, J.R.P.M.; Gorbalenya, A.E.; Lohmann, V.; Van Der Schaar, H.M.; Van Kuppeveld, F.J.M. Modulation of the Host Lipid Landscape to Promote RNA Virus Replication: The Picornavirus Encephalomyocarditis Virus Converges on the Pathway Used by Hepatitis C Virus. PLoS Pathog. 2015, 11, e1005185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, N.-Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.-H.; Takvorian, P.M.; Pau, C.; Van Der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral Reorganization of the Secretory Pathway Generates Distinct Organelles for RNA Replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melia, C.E.; Van Der Schaar, H.M.; Lyoo, H.; Limpens, R.W.; Feng, Q.; Wahedi, M.; Overheul, G.J.; Van Rij, R.P.; Snijder, E.J.; Koster, A.J.; et al. Escaping Host Factor PI4KB Inhibition: Enterovirus Genomic RNA Replication in the Absence of Replication Organelles. Cell Rep. 2017, 21, 587–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roulin, P.S.; Lötzerich, M.; Torta, F.; Tanner, L.B.; Van Kuppeveld, F.J.; Wenk, M.R.; Greber, U.F. Rhinovirus Uses a Phosphatidylinositol 4-Phosphate/Cholesterol Counter-Current for the Formation of Replication Compartments at the ER-Golgi Interface. Cell Host Microbe 2014, 16, 677–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rust, R.C.; Landmann, L.; Gosert, R.; Tang, B.L.; Hong, W.; Hauri, H.-P.; Egger, D.; Bienz, K. Cellular COPII Proteins Are Involved in Production of the Vesicles That Form the Poliovirus Replication Complex. J. Virol. 2001, 75, 9808–9818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suhy, D.A.; Giddings, T.H.; Kirkegaard, K. Remodeling the Endoplasmic Reticulum by Poliovirus Infection and by Individual Viral Proteins: An Autophagy-Like Origin for Virus-Induced Vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef] [Green Version]
- Van Der Schaar, H.M.; Dorobantu, C.M.; Albulescu, L.; Strating, J.R.; Van Kuppeveld, F.J. Fat(al) attraction: Picornaviruses Usurp Lipid Transfer at Membrane Contact Sites to Create Replication Organelles. Trends Microbiol. 2016, 24, 535–546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagy, P.D.; Strating, J.R.P.M.; Van Kuppeveld, F.J.M. Building Viral Replication Organelles: Close Encounters of the Membrane Types. PLoS Pathog. 2016, 12, e1005912. [Google Scholar] [CrossRef]
- Ilnytska, O.; Santiana, M.; Hsu, N.-Y.; Du, W.-L.; Chen, Y.-H.; Viktorova, E.G.; Belov, G.; Brinker, A.; Storch, J.; Moore, C.; et al. Enteroviruses Harness the Cellular Endocytic Machinery to Remodel the Host Cell Cholesterol Landscape for Effective Viral Replication. Cell Host Microbe 2013, 14, 281–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa-Sasaki, K.; Sasaki, J.; Taniguchi, K. A Complex Comprising Phosphatidylinositol 4-Kinase III, ACBD3, and Aichi Virus Proteins Enhances Phosphatidylinositol 4-Phosphate Synthesis and Is Critical for Formation of the Viral Replication Complex. J. Virol. 2014, 88, 6586–6598. [Google Scholar] [CrossRef] [Green Version]
- Albulescu, L.; Wubbolts, R.; Van Kuppeveld, F.J.M.; Strating, J.R.P.M. Cholesterol shuttling is important for RNA replication of coxsackievirus B3 and encephalomyocarditis virus. Cell. Microbiol. 2015, 17, 1144–1156. [Google Scholar] [CrossRef]
- Banerjee, S.; Aponte-Diaz, D.; Yeager, C.; Sharma, S.D.; Ning, G.; Oh, H.S.; Han, Q.; Umeda, M.; Hara, Y.; Wang, R.Y.L.; et al. Hijacking of multiple phospholipid biosynthetic pathways and induction of membrane biogenesis by a picornaviral 3CD protein. PLoS Pathog. 2018, 14, e1007086. [Google Scholar] [CrossRef] [Green Version]
- Antonny, B.; Bigay, J.; Mesmin, B. The Oxysterol-Binding Protein Cycle: Burning Off PI(4)P to Transport Cholesterol. Annu. Rev. Biochem. 2018, 87, 809–837. [Google Scholar] [CrossRef]
- Mesmin, B.; Bigay, J.; Von Filseck, J.M.; Lacas-Gervais, S.; Drin, G.; Antonny, B. A Four-Step Cycle Driven by PI(4)P Hydrolysis Directs Sterol/PI(4)P Exchange by the ER-Golgi Tether OSBP. Cell 2013, 155, 830–843. [Google Scholar] [CrossRef] [Green Version]
- Arita, M. Phosphatidylinositol-4 kinase III beta and oxysterol-binding protein accumulate unesterified cholesterol on poliovirus-induced membrane structure. Microbiol. Immunol. 2014, 58, 239–256. [Google Scholar] [CrossRef]
- Ishikawa-Sasaki, K.; Nagashima, S.; Taniguchi, K.; Sasaki, J. Model of OSBP-Mediated Cholesterol Supply to Aichi Virus RNA Replication Sites Involving Protein-Protein Interactions among Viral Proteins, ACBD3, OSBP, VAP-A/B, and SAC1. J. Virol. 2018, 92, e01952-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Nagy, P.D. Sterol Binding by the Tombusviral Replication Proteins Is Essential for Replication in Yeast and Plants. J. Virol. 2017, 91, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berryman, S.; Moffat, K.; Harak, C.; Lohmann, V.; Jackson, T. Foot-and-mouth disease virus replicates independently of phosphatidylinositol 4-phosphate and type III phosphatidylinositol 4-kinases. J. Gen. Virol. 2016, 97, 1841–1852. [Google Scholar] [CrossRef] [Green Version]
- Esser-Nobis, K.; Harak, C.; Schult, P.; Kusov, Y.; Lohmann, V. Novel perspectives for hepatitis A virus therapy revealed by comparative analysis of hepatitis C virus and hepatitis A virus RNA replication. Hepatology 2015, 62, 397–408. [Google Scholar] [CrossRef]
- Spickler, C.; Lippens, J.; Laberge, M.K.; Desmeules, S.; Bellavance, É.; Garneau, M.; Guo, T.; Hucke, O.; Leyssen, P.; Neyts, J.; et al. Phosphatidylinositol 4-Kinase III Beta Is Essential for Replication of Human Rhinovirus and Its Inhibition Causes a Lethal PhenotypeIn Vivo. Antimicrob. Agents Chemother. 2013, 57, 3358–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, X.; Lei, X.; Zhang, Z.; Ma, Y.; Qi, J.; Wu, C.; Xiao, Y.; Li, L.; He, B.; Wang, J. Enterovirus 3A Facilitates Viral Replication by Promoting Phosphatidylinositol 4-Kinase IIIβ–ACBD3 Interaction. J. Virol. 2017, 91, e00791-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, J.; Nakatsu, F.; Baskin, J.M.; De Camilli, P. Plasticity of PI 4 KIII α interactions at the plasma membrane. EMBO Rep. 2015, 16, 312–320. [Google Scholar] [CrossRef]
- Clayton, E.L.; Minogue, S.; Waugh, M.G. Mammalian phosphatidylinositol 4-kinases as modulators of membrane trafficking and lipid signaling networks. Prog. Lipid Res. 2013, 52, 294–304. [Google Scholar] [CrossRef] [Green Version]
- Daboussi, L.; Costaguta, G.; Ghukasyan, R.; Payne, G.S. Conserved role for Gga proteins in phosphatidylinositol 4-kinase localization to the trans-Golgi network. Proc. Natl. Acad. Sci. USA 2017, 114, 3433–3438. [Google Scholar] [CrossRef] [Green Version]
- De Graaf, P.; Zwart, W.T.; Van Dijken, R.A.; Deneka, M.; Schulz, T.K.; Geijsen, N.; Coffer, P.J.; Gadella, B.M.; Verkleij, A.J.; Van Der Sluijs, P.; et al. Phosphatidylinositol 4-Kinaseβ Is Critical for Functional Association of rab11 with the Golgi Complex. Mol. Biol. Cell 2004, 15, 2038–2047. [Google Scholar] [CrossRef]
- Hausser, A.; Link, G.; Hoene, M.; Russo, C.; Selchow, O.; Pfizenmaier, K. Phospho-specific binding of 14-3-3 proteins to phosphatidylinositol 4-kinase III β protects from dephosphorylation and stabilizes lipid kinase activity. J. Cell Sci. 2006, 119, 3613–3621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haynes, L.P.; Thomas, G.M.H.; Burgoyne, R.D. Interaction of Neuronal Calcium Sensor-1 and ADP-ribosylation Factor 1 Allows Bidirectional Control of Phosphatidylinositol 4-Kinase β and trans-Golgi Network-Plasma Membrane Traffic. J. Biol. Chem. 2005, 280, 6047–6054. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, J.; Ishikawa, K.; Arita, M.; Taniguchi, K. ACBD3-mediated recruitment of PI4KB to picornavirus RNA replication sites. EMBO J. 2011, 31, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Várnai, P.; Tuymetova, G.; Balla, A.; Tóth, Z.E.; Oker-Blom, C.; Roder, J.; Jeromin, A.; Balla, T. Interaction of Neuronal Calcium Sensor-1 (NCS-1) with Phosphatidylinositol 4-Kinase β Stimulates Lipid Kinase Activity and Affects Membrane Trafficking in COS-7 Cells. J. Biol. Chem. 2001, 276, 40183–40189. [Google Scholar] [CrossRef] [Green Version]
- Godi, A.; Pertile, P.; Meyers, R.; Marra, P.; Di Tullio, G.; Iurisci, C.; Luini, A.; Corda, D.; De Matteis, M.A. ARF mediates recruitment of PtdIns-4-OH kinase-β and stimulates synthesis of PtdIns(4,5)P2 on the Golgi complex. Nat. Cell Biol. 1999, 1, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Cuconati, A.; Molla, A.; Wimmer, E. Brefeldin A Inhibits Cell-Free, De Novo Synthesis of Poliovirus. J. Virol. 1998, 72, 6456–6464. [Google Scholar] [CrossRef] [Green Version]
- Maynell, L.A.; Kirkegaard, K.; Klymkowsky, M.W. Inhibition of poliovirus RNA synthesis by brefeldin A. J. Virol. 1992, 66, 1985–1994. [Google Scholar] [CrossRef] [Green Version]
- Donaldson, J.G.; Finazzi, D.; Klausner, R.D. Brefeldin A inhibits Golgi membrane-catalysed exchange of guanine nucleotide onto ARF protein. Nat. Cell Biol. 1992, 360, 350–352. [Google Scholar] [CrossRef]
- Helms, J.B.; Rothman, J.E. Inhibition by brefeldin A of a Golgi membrane enzyme that catalyses exchange of guanine nucleotide bound to ARF. Nat. Cell Biol. 1992, 360, 352–354. [Google Scholar] [CrossRef]
- Klausner, R.D.; Donaldson, J.G.; Lippincott-Schwartz, J. Brefeldin A: Insights into the control of membrane traffic and organelle structure. J. Cell Biol. 1992, 116, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Belov, G.A.; Feng, Q.; Nikovics, K.; Jackson, C.L.; Ehrenfeld, E. A Critical Role of a Cellular Membrane Traffic Protein in Poliovirus RNA Replication. PLoS Pathog. 2008, 4, e1000216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lanke, K.H.W.; Van Der Schaar, H.M.; Belov, G.A.; Feng, Q.; Duijsings, D.; Jackson, C.L.; Ehrenfeld, E.; Van Kuppeveld, F.J.M. GBF1, a Guanine Nucleotide Exchange Factor for Arf, Is Crucial for Coxsackievirus B3 RNA Replication. J. Virol. 2009, 83, 11940–11949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viktorova, E.G.; Gabaglio, S.; Meissner, J.M.; Lee, E.; Moghimi, S.; Sztul, E.; Belov, G.A. A Redundant Mechanism of Recruitment Underlies the Remarkable Plasticity of the Requirement of Poliovirus Replication for the Cellular ArfGEF GBF1. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Belov, G.A.; Altan-Bonnet, N.; Kovtunovych, G.; Jackson, C.L.; Lippincott-Schwartz, J.; Ehrenfeld, E. Hijacking Components of the Cellular Secretory Pathway for Replication of Poliovirus RNA. J. Virol. 2006, 81, 558–567. [Google Scholar] [CrossRef] [Green Version]
- Ferlin, J.; Farhat, R.; Belouzard, S.; Cocquerel, L.; Bertin, A.; Hober, D.; Dubuisson, J.; Rouillé, Y. Investigation of the role of GBF1 in the replication of positive-sense single-stranded RNA viruses. J. Gen. Virol. 2018, 99, 1086–1096. [Google Scholar] [CrossRef]
- Wang, J.; Du, J.; Jin, Q. Class I ADP-Ribosylation Factors Are Involved in Enterovirus 71 Replication. PLoS ONE 2014, 9, e99768. [Google Scholar] [CrossRef]
- Belov, G.A.; Fogg, M.H.; Ehrenfeld, E. Poliovirus Proteins Induce Membrane Association of GTPase ADP-Ribosylation Factor. J. Virol. 2005, 79, 7207–7216. [Google Scholar] [CrossRef] [Green Version]
- Dorobantu, C.M.; Ford-Siltz, L.A.; Sittig, S.P.; Lanke, K.H.W.; Belov, G.A.; Van Kuppeveld, F.J.M.; Van Der Schaar, H.M. GBF1- and ACBD3-Independent Recruitment of PI4KIIIβ to Replication Sites by Rhinovirus 3A Proteins. J. Virol. 2015, 89, 1913–1918. [Google Scholar] [CrossRef] [Green Version]
- Irurzun, A.; Perez, L.; Carrasco, L. Involvement of membrane traffic in the replication of poliovirus genomes: Effects of brefeldin A. Virology 1992, 191, 166–175. [Google Scholar] [CrossRef]
- Dorobantu, C.M.; Van Der Schaar, H.M.; Ford, L.A.; Strating, J.R.P.M.; Ulferts, R.; Fang, Y.; Belov, G.; Van Kuppeveld, F.J.M.; Sandri-Goldin, R.M. Recruitment of PI4KIII to Coxsackievirus B3 Replication Organelles Is Independent of ACBD3, GBF1, and Arf1. J. Virol. 2013, 88, 2725–2736. [Google Scholar] [CrossRef] [Green Version]
- Belov, G.A.; Kovtunovych, G.; Jackson, C.L.; Ehrenfeld, E. Poliovirus replication requires the N-terminus but not the catalytic Sec7 domain of ArfGEF GBF1. Cell. Microbiol. 2010, 12, 1463–1479. [Google Scholar] [CrossRef] [PubMed]
- Altan-Bonnet, N.; Balla, T. Phosphatidylinositol 4-kinases: Hostages harnessed to build panviral replication platforms. Trends Biochem. Sci. 2012, 37, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Golinelli-Cohen, M.; Smirnova, E.; Jackson, C.L. A COPI coat subunit interacts directly with an early-Golgi localized Arf exchange factor. EMBO Rep. 2009, 10, 58–64. [Google Scholar] [CrossRef] [Green Version]
- Belov, G.A.; Habbersett, C.; Franco, D.; Ehrenfeld, E. Activation of Cellular Arf GTPases by Poliovirus Protein 3CD Correlates with Virus Replication. J. Virol. 2007, 81, 9259–9267. [Google Scholar] [CrossRef] [Green Version]
- Van Der Linden, L.; Van Der Schaar, H.M.; Lanke, K.H.W.; Neyts, J.; Van Kuppeveld, F.J.M. Differential Effects of the Putative GBF1 Inhibitors Golgicide A and AG1478 on Enterovirus Replication. J. Virol. 2010, 84, 7535–7542. [Google Scholar] [CrossRef] [Green Version]
- Lowery, J.; Szul, T.; Styers, M.L.; Holloway, Z.G.; Oorschot, V.; Klumperman, J.; Sztul, E. The Sec7 Guanine Nucleotide Exchange Factor GBF1 Regulates Membrane Recruitment of BIG1 and BIG2 Guanine Nucleotide Exchange Factors to the Trans-Golgi Network (TGN). J. Biol. Chem. 2013, 288, 11532–11545. [Google Scholar] [CrossRef] [Green Version]
- Moghimi, S.; Viktorova, E.; Zimina, A.; Szul, T.; Sztul, E.; Belov, G.A. Enterovirus Infection Induces Massive Recruitment of All Isoforms of Small Cellular Arf GTPases to the Replication Organelles. J. Virol. 2020, 95, 95. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.S.; Banerjee, S.; Aponte-Diaz, D.; Sharma, S.D.; Aligo, J.; Lodeiro, M.F.; Ning, G.; Sharma, R.; Arnold, J.J.; Cameron, C.E. Multiple poliovirus-induced organelles suggested by comparison of spatiotemporal dynamics of membranous structures and phosphoinositides. PLoS Pathog. 2018, 14, e1007036. [Google Scholar] [CrossRef] [Green Version]
- Cameron, C.E.; Oh, H.S.; Moustafa, I.M. Expanding knowledge of P3 proteins in the poliovirus lifecycle. Futur. Microbiol. 2010, 5, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Crotty, S.; Saleh, M.-C.; Gitlin, L.; Beske, O.; Andino, R. The Poliovirus Replication Machinery Can Escape Inhibition by an Antiviral Drug That Targets a Host Cell Protein. J. Virol. 2004, 78, 3378–3386. [Google Scholar] [CrossRef] [Green Version]
- Arita, M.; Kojima, H.; Nagano, T.; Okabe, T.; Wakita, T.; Shimizu, H. Phosphatidylinositol 4-Kinase III Beta Is a Target of Enviroxime-Like Compounds for Antipoliovirus Activity. J. Virol. 2010, 85, 2364–2372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Der Schaar, H.M.; Van Der Linden, L.; Lanke, K.H.W.; Strating, J.R.P.M.; Pürstinger, G.; De Vries, E.; Haan, C.A.M.D.; Neyts, J.; Van Kuppeveld, F.J.M. Coxsackievirus mutants that can bypass host factor PI4KIIIβ and the need for high levels of PI4P lipids for replication. Cell Res. 2012, 22, 1576–1592. [Google Scholar] [CrossRef] [Green Version]
- Van Der Schaar, H.M.; Leyssen, P.; Thibaut, H.J.; De Palma, A.; Van Der Linden, L.; Lanke, K.H.W.; Lacroix, C.; Verbeken, E.; Conrath, K.; MacLeod, A.M.; et al. A Novel, Broad-Spectrum Inhibitor of Enterovirus Replication That Targets Host Cell Factor Phosphatidylinositol 4-Kinase IIIβ. Antimicrob. Agents Chemother. 2013, 57, 4971–4981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strating, J.R.; Van Der Linden, L.; Albulescu, L.; Bigay, J.; Arita, M.; Delang, L.; Leyssen, P.; Van Der Schaar, H.M.; Lanke, K.H.; Thibaut, H.J.; et al. Itraconazole Inhibits Enterovirus Replication by Targeting the Oxysterol-Binding Protein. Cell Rep. 2015, 10, 600–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albulescu, L.; Strating, J.R.; Thibaut, H.J.; Van Der Linden, L.; Shair, M.D.; Neyts, J.; Van Kuppeveld, F.J. Broad-range inhibition of enterovirus replication by OSW-1, a natural compound targeting OSBP. Antivir. Res. 2015, 117, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Arita, M. Mechanism of Poliovirus Resistance to Host Phosphatidylinositol-4 Kinase III β Inhibitor. ACS Infect. Dis. 2016, 2, 140–148. [Google Scholar] [CrossRef]
- Albulescu, L.; Bigay, J.; Biswas, B.; Weber-Boyvat, M.; Dorobantu, C.M.; Delang, L.; Van Der Schaar, H.M.; Jung, Y.-S.; Neyts, J.; Olkkonen, V.M.; et al. Uncovering oxysterol-binding protein (OSBP) as a target of the anti-enteroviral compound TTP-8307. Antivir. Res. 2017, 140, 37–44. [Google Scholar] [CrossRef]
- Heinz, B.A.; Vance, L.M. Sequence determinants of 3A-mediated resistance to enviroxime in rhinoviruses and enteroviruses. J. Virol. 1996, 70, 4854–4857. [Google Scholar] [CrossRef] [Green Version]
- Arita, M.; Wakita, T.; Shimizu, H. Cellular kinase inhibitors that suppress enterovirus replication have a conserved target in viral protein 3A similar to that of enviroxime. J. Gen. Virol. 2009, 90, 1869–1879. [Google Scholar] [CrossRef]
- Lyoo, H.; Dorobantu, C.M.; Van Der Schaar, H.M.; Van Kuppeveld, F.J. Modulation of proteolytic polyprotein processing by coxsackievirus mutants resistant to inhibitors targeting phosphatidylinositol-4-kinase IIIβ or oxysterol binding protein. Antivir. Res. 2017, 147, 86–90. [Google Scholar] [CrossRef]
- Arita, M.; Bigay, J. Poliovirus Evolution toward Independence from the Phosphatidylinositol-4 Kinase III β/Oxysterol-Binding Protein Family I Pathway. ACS Infect. Dis. 2019, 5, 962–973. [Google Scholar] [CrossRef] [PubMed]
- Dorobantu, C.M.; Albulescu, L.; Lyoo, H.; Van Kampen, M.; De Francesco, R.; Lohmann, V.; Harak, C.; Van Der Schaar, H.M.; Strating, J.R.P.M.; Gorbalenya, A.E.; et al. Mutations in Encephalomyocarditis Virus 3A Protein Uncouple the Dependency of Genome Replication on Host Factors Phosphatidylinositol 4-Kinase IIIα and Oxysterol-Binding Protein. mSphere 2016, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burke, J.E.; Inglis, A.J.; Perisic, O.; Masson, G.R.; McLaughlin, S.H.; Rutaganira, F.; Shokat, K.M.; Williams, R.L. Structures of PI4KIII complexes show simultaneous recruitment of Rab11 and its effectors. Science 2014, 344, 1035–1038. [Google Scholar] [CrossRef] [Green Version]
- Valente, C.; Turacchio, G.; Mariggiò, S.; Pagliuso, A.; Gaibisso, R.; Di Tullio, G.; Santoro, M.; Formiggini, F.; Spanò, S.; Piccini, D.; et al. A 14-3-3γ dimer-based scaffold bridges CtBP1-S/BARS to PI(4)KIIIβ to regulate post-Golgi carrier formation. Nat. Cell Biol. 2012, 14, 343–354. [Google Scholar] [CrossRef]
- Klima, M.; Tóth, D.J.; Hexnerova, R.; Baumlova, A.; Chalupska, D.; Tykvart, J.; Rezabkova, L.; Sengupta, N.; Man, P.; Dubankova, A.; et al. Structural insights and in vitro reconstitution of membrane targeting and activation of human PI4KB by the ACBD3 protein. Sci. Rep. 2016, 6, 23641. [Google Scholar] [CrossRef]
- Petrosyan, A. Unlocking Golgi: Why Does Morphology Matter? Biochem. (Moscow) 2019, 84, 1490–1501. [Google Scholar] [CrossRef]
- Sohda, M.; Misumi, Y.; Yamamoto, A.; Yano, A.; Nakamura, N.; Ikehara, Y. Identification and Characterization of a Novel Golgi Protein, GCP60, That Interacts with the Integral Membrane Protein Giantin. J. Biol. Chem. 2001, 276, 45298–45306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greninger, A.L.; Knudsen, G.M.; Betegon, M.; Burlingame, A.L.; DeRisi, J.L. ACBD3 Interaction with TBC1 Domain 22 Protein Is Differentially Affected by Enteroviral and Kobuviral 3A Protein Binding. mBio 2013, 4, e00098-13. [Google Scholar] [CrossRef] [Green Version]
- Téoulé, F.; Brisac, C.; Pelletier, I.; Vidalain, P.-O.; Jégouic, S.; Mirabelli, C.; Bessaud, M.; Combelas, N.; Autret, A.; Tangy, F.; et al. The Golgi Protein ACBD3, an Interactor for Poliovirus Protein 3A, Modulates Poliovirus Replication. J. Virol. 2013, 87, 11031–11046. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.S.; Lee, K.; Kim, S.-J.; Cho, S.; Shin, H.J.; Kim, C.; Kim, J.-S. Arrayed CRISPR screen with image-based assay reliably uncovers host genes required for coxsackievirus infection. Genome Res. 2018, 28, 859–868. [Google Scholar] [CrossRef] [Green Version]
- Lei, X.; Xiao, X.; Zhang, Z.; Ma, Y.; Qi, J.; Wu, C.; Xiao, Y.; Zhou, Z.; He, B.; Wang, J. The Golgi protein ACBD3 facilitates Enterovirus 71 replication by interacting with 3A. Sci. Rep. 2017, 7, 44592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyoo, H.; Van Der Schaar, H.M.; Dorobantu, C.M.; Rabouw, H.H.; Strating, J.R.P.M.; Van Kuppeveld, F.J.M. ACBD3 Is an Essential Pan-enterovirus Host Factor That Mediates the Interaction between Viral 3A Protein and Cellular Protein PI4KB. mBio 2019, 10, e02742-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klima, M.; Chalupska, D.; Różycki, B.; Humpolickova, J.; Rezabkova, L.; Silhan, J.; Baumlova, A.; Dubankova, A.; Boura, E. Kobuviral Non-structural 3A Proteins Act as Molecular Harnesses to Hijack the Host ACBD3 Protein. Structure 2017, 25, 219–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islinger, M.; Costello, J.L.; Kors, S.; Soupene, E.; Levine, T.P.; Kuypers, F.A.; Schrader, M. The diversity of ACBD proteins—From lipid binding to protein modulators and organelle tethers. Biochim. Biophys. Acta BBA Bioenerg. 2020, 1867, 118675. [Google Scholar] [CrossRef]
- Yue, X.; Qian, Y.; Gim, B.; Lee, I. Acyl-CoA-Binding Domain-Containing 3 (ACBD3; PAP7; GCP60): A Multi-Functional Membrane Domain Organizer. Int. J. Mol. Sci. 2019, 20, 2028. [Google Scholar] [CrossRef] [Green Version]
- McPhail, J.A.; Ottosen, E.H.; Jenkins, M.L.; Burke, J.E. The Molecular Basis of Aichi Virus 3A Protein Activation of Phosphatidylinositol 4 Kinase IIIβ, PI4KB, through ACBD3. Struct. 2017, 25, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Wei, Z.; Zhang, M.; Li, C.; Huang, W.; Fan, Y.; Guo, J.; Khater, M.; Fukuda, M.; Dong, Z.; Hu, G.; et al. Specific TBC Domain-Containing Proteins Control the ER-Golgi-Plasma Membrane Trafficking of GPCRs. Cell Rep. 2019, 28, 554–566.e4. [Google Scholar] [CrossRef] [PubMed]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef] [PubMed]
- Thiam, A.R.; Farese, R.V., Jr.; Walther, T.C. The biophysics and cell biology of lipid droplets. Nat. Rev. Mol. Cell Biol. 2013, 14, 775–786. [Google Scholar] [CrossRef] [Green Version]
- Laufman, O.; Perrino, J.; Andino, R. Viral Generated Inter-Organelle Contacts Redirect Lipid Flux for Genome Replication. Cell 2019, 178, 275–289.e16. [Google Scholar] [CrossRef]
- Guan, H.; Tian, J.; Zhang, C.; Qin, B.; Cui, S. Crystal structure of a soluble fragment of poliovirus 2CATPase. PLoS Pathog. 2018, 14, e1007304. [Google Scholar] [CrossRef] [PubMed]
- Stevanovic, A.; Thiele, C. Monotopic topology is required for lipid droplet targeting of ancient ubiquitous protein 1. J. Lipid Res. 2013, 54, 503–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellong, E.N.; Soni, K.G.; Bui, Q.-T.; Sougrat, R.; Golinelli-Cohen, M.-P.; Jackson, C.L. Interaction between the Triglyceride Lipase ATGL and the Arf1 Activator GBF1. PLoS ONE 2011, 6, e21889. [Google Scholar] [CrossRef] [Green Version]
- Soni, K.G.; Mardones, G.A.; Sougrat, R.; Smirnova, E.; Jackson, C.L.; Bonifacino, J.S. Coatomer-dependent protein delivery to lipid droplets. J. Cell Sci. 2009, 122, 1834–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Claude, A.; Chun, J.; Shields, D.J.; Presley, J.F.; Melançon, P. GBF1, a cis-Golgi and VTCs-localized ARF-GEF, is implicated in ER-to-Golgi protein traffic. J. Cell Sci. 2006, 119, 3743–3753. [Google Scholar] [CrossRef] [Green Version]
- Belov, G.A.; Van Kuppeveld, F.J. Lipid Droplets Grease Enterovirus Replication. Cell Host Microbe 2019, 26, 149–151. [Google Scholar] [CrossRef] [Green Version]
- Medina, G.N.; Segundo, F.D.-S.; Stenfeldt, C.; Arzt, J.; Santos, T.D.L. The Different Tactics of Foot-and-Mouth Disease Virus to Evade Innate Immunity. Front. Microbiol. 2018, 9, 2644. [Google Scholar] [CrossRef]
- Lloyd, R.E. Enterovirus Control of Translation and RNA Granule Stress Responses. Viruses 2016, 8, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]





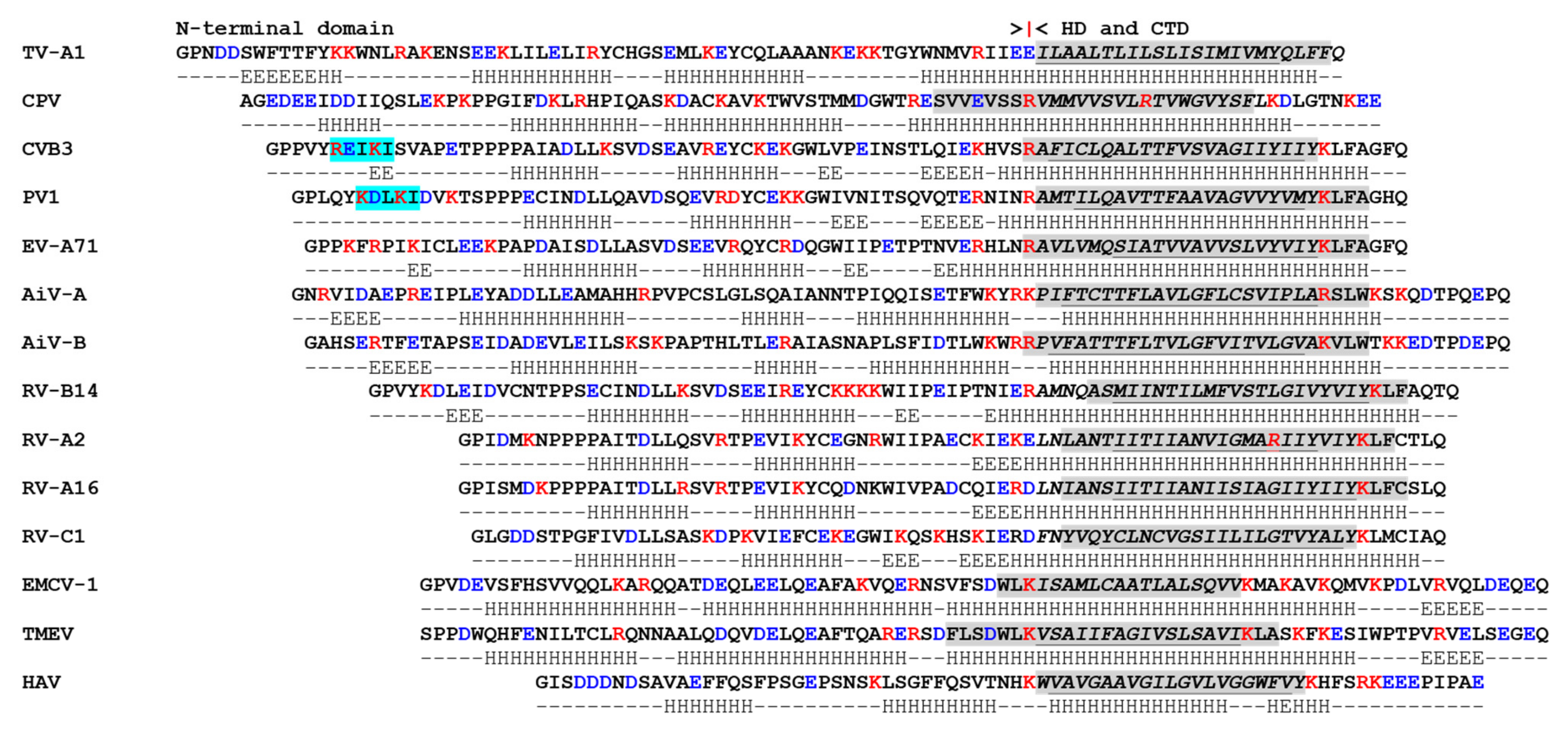{kind=link}
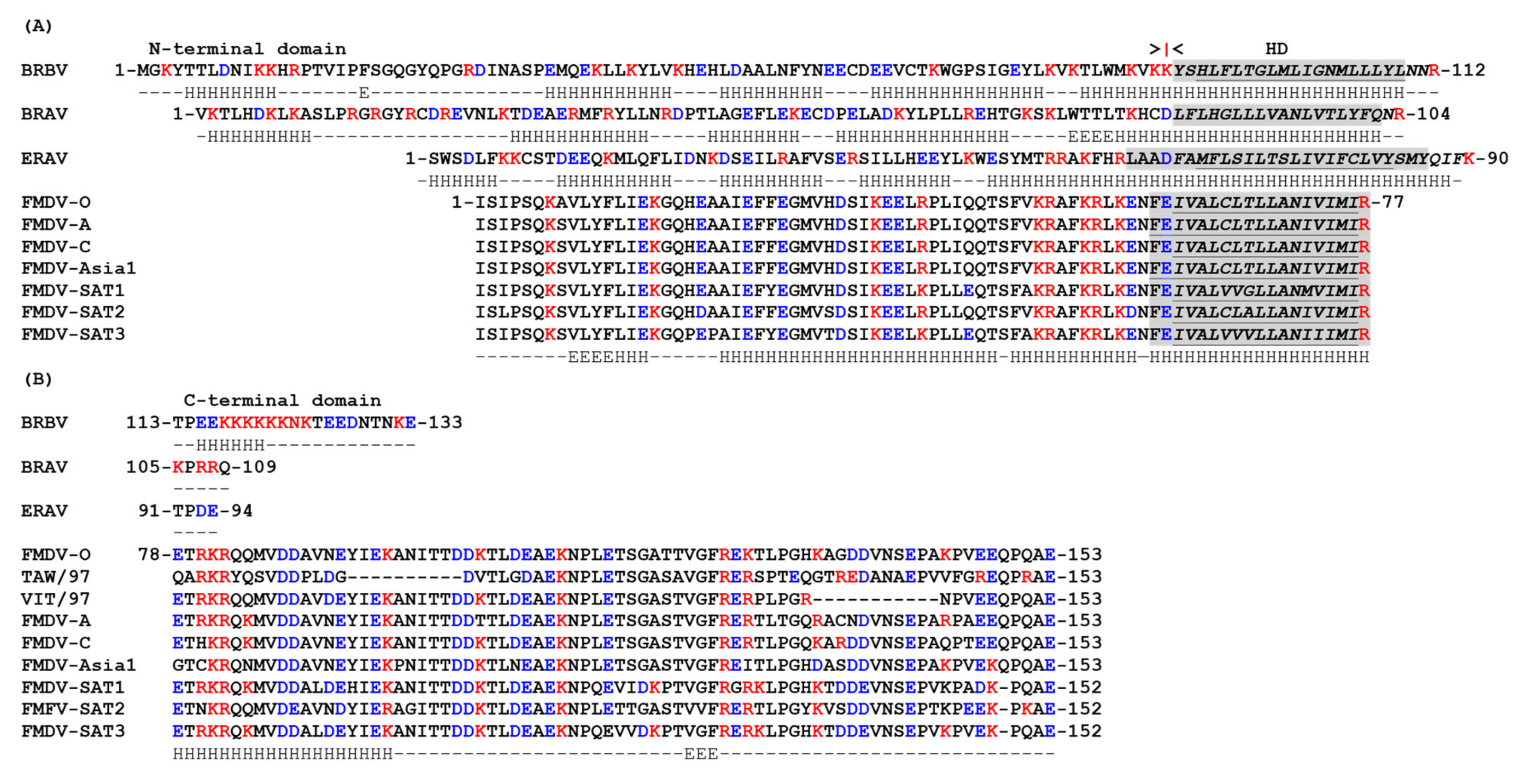{kind=link}
{kind=link}
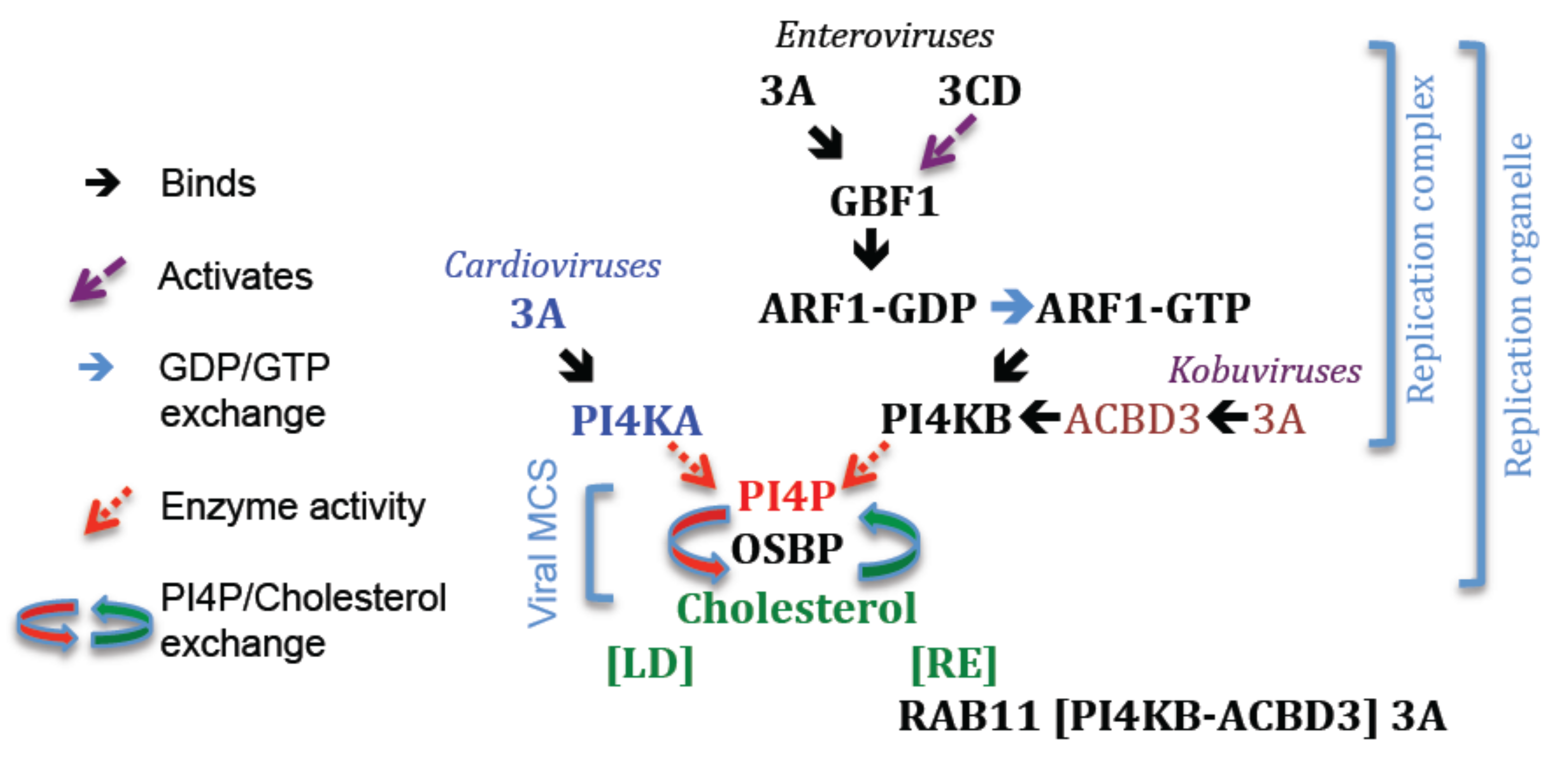{kind=link}
| Picornavirus General Genome Organization: | |||||
|---|---|---|---|---|---|
| 1 x ORF VPg-5’-UTR-IRES -(±L)-1A-1B-1C-1D-2A-2B-2C-3A-3B-3C-3D -3’-UTR-poly(A) | |||||
| Genus | IRES | Genus Specific Differences | |||
| Aphthovirus | II | Lpro | 2Anpgp | 3B13B23B3 | |
| Aalivirus | IV | 1AB | 2A1npgp2A2npgp2A3npgp2A4npgp2A52A6 | ||
| Aquamavirus | IV | 1AB | 2A1npgp2A2 | 3B13B2 | |
| Avihepatovirus | IV | 1AB | 2A1npgp2A2NTPase2A3H-box/NC | ||
| Cardiovirus | II | L | 2Anpgp | ||
| Enterovirus | I | 2Apro | |||
| Kobuvirus | V | L | 1AB | 2AH-box/NC | |
| Limnipivirus | IV | 1AB | 2A1npgp2A2npgp | ||
| Mosavirus | II | L | 2Anpgp | 3B13B2 | |
| Parechovirus | II | 1AB | 2A1npgp2A2H-box/NC | ||
| 2 x ORF VPg-5’-UTR-IRES1-1A-1B-1C-1D-IGR-IRES2-2A-2B-2C-3A-3B-3C-3D -3’-UTR-poly(A) | |||||
| Dicipivirus | II1 / I 2 | ORF1 1A-1B-1C-1D | ORF2 2A-2B-2C | ORF2 3A-3B-3C-3D | |
| Y-2-H [111,112] | M-2-H [110] | M-2-H [109] | |||||
|---|---|---|---|---|---|---|---|
| PV | AiV | ||||||
| 3A | 3AB | 3A | 3AB | 3A | 3AB | ||
| PV 2A | + | – | ND | ND | AiV 2A | + | ND |
| PV 2B | +* | +* | + | + | AiV 2B | + | – |
| PV 2C | +* | +* | + | - | AiV 2C | + | + |
| PV 2BC | – | – | + | + | AiV 2BC | + | + |
| PV 3A | +* | + | + | + | AiV 3A | + | + |
| PV 3B | – | – | ND | ND | AiV 3B | ND | ND |
| PV 3AB | + | + | + | – | AiV 3AB | + | – |
| PV 3C | – | – | ND | ND | AiV 3C | – | + |
| PV 3D | – | +* | ND | ND | AiV 3D | – | – |
| PV 3CD | – | +* | ND | ND | AiV 3CD | – | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jackson, T.; Belsham, G.J. Picornaviruses: A View from 3A. Viruses 2021, 13, 456. https://doi.org/10.3390/v13030456
Jackson T, Belsham GJ. Picornaviruses: A View from 3A. Viruses. 2021; 13(3):456. https://doi.org/10.3390/v13030456
Chicago/Turabian StyleJackson, Terry, and Graham J. Belsham. 2021. "Picornaviruses: A View from 3A" Viruses 13, no. 3: 456. https://doi.org/10.3390/v13030456
APA StyleJackson, T., & Belsham, G. J. (2021). Picornaviruses: A View from 3A. Viruses, 13(3), 456. https://doi.org/10.3390/v13030456