
In the Era of mRNA Vaccines, Is There Any Hope for HIV Functional Cure?

 and
and
Abstract
:1. Introduction
2. Types and Molecular Biology of mRNA Vaccines
3. mRNA Vaccine Delivery Methods
3.1. Naked mRNA
3.2. Dendritic Cell Vaccination
3.3. Formulated mRNA
4. Therapeutic mRNA Vaccines in Clinical Trials: The Experience with HIV
4.1. mRNA DC Vaccination
4.2. Naked mRNA Vaccination
5. Improving mRNA Vaccines for HIV
6. mRNA-Based HIV Functional Cure Strategies
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Meliopoulos, V.A.; Wang, W.; Lin, X.; Stucker, K.M.; Halpin, R.A.; Stockwell, T.B.; Schultz-Cherry, S.; Wentworth, D.E. Reversion of Cold-Adapted Live Attenuated Influenza Vaccine into a Pathogenic Virus. J. Virol. 2016, 90, 8454–8463. [Google Scholar] [CrossRef] [Green Version]
- Baitsch, L.; Baumgaertner, P.; Devêvre, E.; Raghav, S.K.; Legat, A.; Barba, L.; Wieckowski, S.; Bouzourene, H.; Deplancke, B.; Romero, P.; et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Investig. 2011, 121, 2350–2360. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Arévalo, M.T.; Chen, Y.; Chen, S.; Zeng, M. T-cell-mediated cross-strain protective immunity elicited by prime-boost vaccination with a live attenuated influenza vaccine. Int. J. Infect. Dis. 2014, 27, 37–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolff, J.A.; Malone, R.W.; Williams, P.; Chong, W.; Acsadi, G.; Jani, A.; Felgner, P.L. Direct gene transfer into mouse muscle in vivo. Science 1990, 247, 1465–1468. [Google Scholar] [CrossRef]
- Perdiguero, B.; Raman, S.C.; Sánchez-Corzo, C.; Sorzano, C.O.S.; Valverde, J.R.; Esteban, M.; Gómez, C.E. Potent HIV-1-specific CD8 T cell responses induced in mice after priming with a multiepitopic DNA-TMEP and boosting with the HIV vaccine MVA-B. Viruses 2018, 10, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdiguero, B.; Sánchez-Corzo, C.; Sorzano, C.O.S.; Mediavilla, P.; Saiz, L.; Esteban, M.; Gómez, C.E. Induction of broad and polyfunctional HIV-1-specific T cell responses by the multiepitopic protein TMEP-B vectored by MVA virus. Vaccines 2019, 7, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mothe, B.; Manzardo, C.; Sanchez-Bernabeu, A.; Coll, P.; Morón-López, S.; Puertas, M.C.; Rosas-Umbert, M.; Cobarsi, P.; Escrig, R.; Perez-Alvarez, N.; et al. Therapeutic Vaccination Refocuses T-cell Responses Towards Conserved Regions of HIV-1 in Early Treated Individuals (BCN 01 study). EClinicalMedicine 2019, 11, 65–80. [Google Scholar] [CrossRef] [Green Version]
- Mothe, B.; Rosás-Umbert, M.; Coll, P.; Manzardo, C.; Puertas, M.C.; Morón-López, S.; Llano, A.; Miranda, C.; Cedeño, S.; López, M.; et al. HIVconsv Vaccines and Romidepsin in Early-Treated HIV-1-Infected Individuals: Safety, Immunogenicity and Effect on the Viral Reservoir (Study BCN02). Front. Immunol. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Vardas, E.; Stanescu, I.; Leinonen, M.; Ellefsen, K.; Pantaleo, G.; Valtavaara, M.; Ustav, M.; Reijonen, K. Indicators of therapeutic effect in FIT-06, a Phase II trial of a DNA vaccine, GTU®-Multi-HIVB, in untreated HIV-1 infected subjects. Vaccine 2012, 30, 4046–4054. [Google Scholar] [CrossRef]
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA recognition by Toll-like receptors: The impact of nucleoside modification and the evolutionary origin of RNA. Immunity 2005, 23, 165–175. [Google Scholar] [CrossRef] [Green Version]
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of pseudouridine into mRNA yields superior nonimmunogenic vector with increased translational capacity and biological stability. Mol. Ther. 2008, 16, 1833–1840. [Google Scholar] [CrossRef]
- Weide, B.; Pascolo, S.; Scheel, B.; Derhovanessian, E.; Pflugfelder, A.; Eigentler, T.K.; Pawelec, G.; Hoerr, I.; Rammensee, H.; Garbe, C. Direct Injection of Protamine-protected mRNA: Melanoma Patients. J. Immunother. 2009, 32, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Routy, J.P.; Boulassel, M.R.; Yassine-Diab, B.; Nicolette, C.; Healey, D.; Jain, R.; Landry, C.; Yegorov, O.; Tcherepanova, I.; Monesmith, T.; et al. Immunologic activity and safety of autologous HIV RNA-electroporated dendritic cells in HIV-1 infected patients receiving antiretroviral therapy. Clin. Immunol. 2010, 134, 140–147. [Google Scholar] [CrossRef] [Green Version]
- Geall, A.J.; Verma, A.; Otten, G.R.; Shaw, C.A.; Hekele, A.; Banerjee, K.; Cu, Y.; Beard, C.W.; Brito, L.A.; Krucker, T.; et al. Nonviral delivery of self-amplifying RNA vaccines. Proc. Natl. Acad. Sci. USA 2012, 109, 14604–14609. [Google Scholar] [CrossRef] [Green Version]
- Martinon, F.; Krishnan, S.; Lenzen, G.; Magné, R.; Gomard, E.; Guillet, J.-G.; Lévy, J.-P.; Meulien, P. Induction of virus-specific cytotoxic T lymphocytes in vivo by liposome-entrapped mRNA. Eur. J. Immunol. 1993, 23, 1719–1722. [Google Scholar] [CrossRef] [PubMed]
- Boczkowski, D.; Nair, S.K.; Snyder, D.; Gilboa, E. Dendritic cells pulsed with RNA are potent antigen-presenting cells in vitro and in vivo. J. Exp. Med. 1996, 184, 465–472. [Google Scholar] [CrossRef] [Green Version]
- Van Gulck, E.; Vlieghe, E.; Vekemans, M.; Van Tendeloo, V.F.I.; Van De Velde, A.; Smits, E.; Anguille, S.; Cools, N.; Goossens, H.; Mertens, L.; et al. MRNA-based dendritic cell vaccination induces potent antiviral T-cell responses in HIV-1-infected patients. AIDS 2012, 26, F1–F12. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; Secreto, A.J.; Shan, X.; Debonera, F.; Glover, J.; Yi, Y.; Muramatsu, H.; Ni, H.; Mui, B.L.; Tam, Y.K.; et al. Administration of nucleoside-modified mRNA encoding broadly neutralizing antibody protects humanized mice from HIV-1 challenge. Nat. Commun. 2017, 8, 14630. [Google Scholar] [CrossRef] [PubMed]
- De Jong, W.; Aerts, J.; Allard, S.; Brander, C.; Buyze, J.; Florence, E.; Van Gorp, E.; Vanham, G.; Leal, L.; Mothe, B.; et al. IHIVARNA phase IIa, a randomized, placebo-controlled, double-blinded trial to evaluate the safety and immunogenicity of iHIVARNA-01 in chronically HIV-infected patients under stable combined antiretroviral therapy. Trials 2019, 20, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Petsch, B.; Schnee, M.; Vogel, A.B.; Lange, E.; Hoffmann, B.; Voss, D.; Schlake, T.; Thess, A.; Kallen, K.J.; Stitz, L.; et al. Protective efficacy of in vitro synthesized, specific mRNA vaccines against influenza A virus infection. Nat. Biotechnol. 2012, 30, 1210–1216. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines-a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.F.; Hung, C.F.; Hsu, K.F.; Chai, C.Y.; He, L.; Ling, M.; Slater, L.A.; Roden, R.B.S.; Wu, T.C. Enhancement of sindbis virus self-replicating RNA vaccine potency by targeting antigen to endosomal/lysosomal compartments. Hum. Gene Ther. 2001, 12, 235–252. [Google Scholar] [CrossRef] [PubMed]
- Ljungberg, K.; Liljeström, P. Self-replicating alphavirus RNA vaccines. Expert Rev. Vaccines 2014, 14, 177–194. [Google Scholar] [CrossRef]
- Rna, S.; Tews, B.A.; Meyers, G. Self-Replicating RNA Birke. RNA Vaccines 2017, 1499. [Google Scholar] [CrossRef]
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jackson, N.A.C.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. NPJ Vaccines 2020, 5, 3–8. [Google Scholar] [CrossRef]
- Li, Y.; Kiledjian, M. Regulation of mRNA decapping. Wiley Interdiscip. Rev. RNA 2010, 1, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Brito, L.A.; Kommareddy, S.; Maione, D.; Uematsu, Y.; Giovani, C.; Berlanda Scorza, F.; Otten, G.R.; Yu, D.; Mandl, C.W.; Mason, P.W.; et al. Self-Amplifying mRNA Vaccines; Elsevier Ltd.: Amsterdam, The Netherlands, 2015; Volume 89. [Google Scholar]
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174. [Google Scholar] [CrossRef]
- Tanguay, R.L.; Gallie, D.R. Translational efficiency is regulated by the length of the 3′ untranslated region. Mol. Cell. Biol. 1996, 16, 146–156. [Google Scholar] [CrossRef] [Green Version]
- Lima, S.A.; Chipman, L.B.; Nicholson, A.L.; Chen, Y.-H.; Yee, B.A.; Yeo, G.W.; Coller, J.; Pasquinelli, A.E. Short poly(A) tails are a conserved feature of highly expressed genes. Nat. Struct. Mol. Biol. 2017, 24, 1057–1063. [Google Scholar] [CrossRef] [Green Version]
- Kudla, G.; Lipinski, L.; Caffin, F.; Helwak, A.; Zylicz, M. High Guanine and Cytosine Content Increases mRNA Levels in Mammalian Cells. PLoS Biol. 2006, 4, e180. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Huang, C.; Yu, Y.T. Pseudouridine in mRNA: Incorporation, Detection, and Recoding, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2015; Volume 560. [Google Scholar]
- Geall, A.J.; Mandl, C.W.; Ulmer, J.B. RNA: The new revolution in nucleic acid vaccines. Semin. Immunol. 2013, 25, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Weissman, D.; Pardi, N.; Muramatsu, H.; Karikó, K. HPLC Purification of In Vitro Transcribed Long RNA. In Synthetic Messenger RNA and Cell Metabolism Modulation; Humana Press: Totowa, NJ, USA, 2013; pp. 43–54. [Google Scholar]
- Karikó, K.; Muramatsu, H.; Ludwig, J.; Weissman, D. Generating the optimal mRNA for therapy: HPLC purification eliminates immune activation and improves translation of nucleoside-modified, protein-encoding mRNA. Nucleic Acids Res. 2011, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Romani, B.; Kavyanifard, A.; Allahbakhshi, E. Antibody production by in vivo RNA transfection. Sci. Rep. 2017, 7, 10863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weissman, D.; Ni, H.; Scales, D.; Dude, A.; Capodici, J.; McGibney, K.; Abdool, A.; Isaacs, S.N.; Cannon, G.; Karikó, K. HIV Gag mRNA Transfection of Dendritic Cells (DC) Delivers Encoded Antigen to MHC Class I and II Molecules, Causes DC Maturation, and Induces a Potent Human In Vitro Primary Immune Response. J. Immunol. 2000, 165, 4710–4717. [Google Scholar] [CrossRef] [Green Version]
- Corr, M.; von Damm, A.; Lee, D.J.; Tighe, H. In vivo priming by DNA injection occurs predominantly by antigen transfer. J. Immunol. 1999, 163, 4721–4727. [Google Scholar] [PubMed]
- Shirota, H.; Petrenko, L.; Hong, C.; Klinman, D.M. Potential of Transfected Muscle Cells to Contribute to DNA Vaccine Immunogenicity. J. Immunol. 2007, 179, 329–336. [Google Scholar] [CrossRef] [Green Version]
- Kowalczyk, A.; Doener, F.; Zanzinger, K.; Noth, J.; Baumhof, P.; Fotin-Mleczek, M.; Heidenreich, R. Self-adjuvanted mRNA vaccines induce local innate immune responses that lead to a potent and boostable adaptive immunity. Vaccine 2016, 34, 3882–3893. [Google Scholar] [CrossRef] [PubMed]
- Bertheloot, D.; Naumovski, A.L.; Langhoff, P.; Horvath, G.L.; Jin, T.; Xiao, T.S.; Garbi, N.; Agrawal, S.; Kolbeck, R.; Latz, E. RAGE Enhances TLR Responses through Binding and Internalization of RNA. J. Immunol. 2016, 197, 4118–4126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, M.; Li, M.; Zhang, Z.; Gong, T.; Sun, X. Induction of HIV-1 gag specific immune responses by cationic micelles mediated delivery of gag mRNA. Drug Deliv. 2016, 23, 2596–2607. [Google Scholar] [CrossRef]
- Fan, Y.-N.; Li, M.; Luo, Y.-L.; Chen, Q.; Wang, L.; Zhang, H.-B.; Shen, S.; Gu, Z.; Wang, J. Cationic lipid-assisted nanoparticles for delivery of mRNA cancer vaccine. Biomater. Sci. 2018, 6, 3009–3018. [Google Scholar] [CrossRef]
- Lou, G.; Anderluzzi, G.; Schmidt, S.T.; Woods, S.; Gallorini, S.; Brazzoli, M.; Giusti, F.; Ferlenghi, I.; Johnson, R.N.; Roberts, C.W.; et al. Delivery of self-amplifying mRNA vaccines by cationic lipid nanoparticles: The impact of cationic lipid selection. J. Control. Release 2020, 325, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Kreiter, S.; Selmi, A.; Diken, M.; Koslowski, M.; Britten, C.M.; Huber, C.; Türeci, Ö.; Sahin, U. Intranodal Vaccination with Naked Antigen-Encoding RNA Elicits Potent Prophylactic and Therapeutic Antitumoral Immunity. Cancer Res. 2010, 70, 9031–9040. [Google Scholar] [CrossRef] [Green Version]
- Guardo, A.C.; Joe, P.T.; Miralles, L.; Bargalló, M.E.; Mothe, B.; Krasniqi, A.; Heirman, C.; García, F.; Thielemans, K.; Brander, C.; et al. Preclinical evaluation of an mRNA HIV vaccine combining rationally selected antigenic sequences and adjuvant signals (HTI-TriMix). Aids 2017, 31, 321–332. [Google Scholar] [CrossRef] [PubMed]
- Gilboa, E.; Vieweg, J. Cancer immunotherapy with mRNA-transfected dendritic cells. Immunol. Rev. 2004, 199, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Bol, K.F.; Figdor, C.G.; Aarntzen, E.H.; Welzen, M.E.; van Rossum, M.M.; Blokx, W.A.; van de Rakt, M.W.; Scharenborg, N.M.; de Boer, A.J.; Pots, J.M.; et al. Intranodal vaccination with mRNA-optimized dendritic cells in metastatic melanoma patients. Oncoimmunology 2015, 4, e1019197. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, M.; de Bruin, M.G.M.; Seuberlich, T.; Balmelli, C.; Pascolo, S.; Ruggli, N.; Wienhold, D.; Tratschin, J.D.; McCullough, K.C.; Summerfield, A. Identification of classical swine fever virus protein E2 as a target for cytotoxic T cells by using mRNA-transfected antigen-presenting cells. J. Gen. Virol. 2005, 86, 2525–2534. [Google Scholar] [CrossRef] [PubMed]
- Hobo, W.; Novobrantseva, T.I.; Fredrix, H.; Wong, J.; Milstein, S.; Epstein-Barash, H.; Liu, J.; Schaap, N.; van der Voort, R.; Dolstra, H. Improving dendritic cell vaccine immunogenicity by silencing PD-1 ligands using siRNA-lipid nanoparticles combined with antigen mRNA electroporation. Cancer Immunol. Immunother. 2013, 62, 285–297. [Google Scholar] [CrossRef]
- Melhem, N.M.; Liu, X.D.; Boczkowski, D.; Gilboa, E.; Barratt-Boyes, S.M. Robust CD4+ and CD8+ T cell responses to SIV using mRNA-transfected DC expressing autologous viral Ag. Eur. J. Immunol. 2007, 37, 2164–2173. [Google Scholar] [CrossRef]
- Romain, G.; van Gulck, E.; Epaulard, O.; Oh, S.; Li, D.; Zurawski, G.; Zurawski, S.; Cosma, A.; Adam, L.; Chapon, C.; et al. CD34-derived dendritic cells transfected ex vivo with HIV-Gag mRNA induce polyfunctional T-cell responses in nonhuman primates. Eur. J. Immunol. 2012, 42, 2019–2030. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhao, M.; Fu, Y.; Li, Y.; Gong, T.; Zhang, Z.; Sun, X. Enhanced intranasal delivery of mRNA vaccine by overcoming the nasal epithelial barrier via intra-and paracellular pathways. J. Control. Release 2016, 228, 9–19. [Google Scholar] [CrossRef]
- Udhayakumar, V.K.; De Beuckelaer, A.; McCaffrey, J.; McCrudden, C.M.; Kirschman, J.L.; Vanover, D.; Van Hoecke, L.; Roose, K.; Deswarte, K.; De Geest, B.G.; et al. Arginine-Rich Peptide-Based mRNA Nanocomplexes Efficiently Instigate Cytotoxic T Cell Immunity Dependent on the Amphipathic Organization of the Peptide. Adv. Healthc. Mater. 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Persano, S.; Guevara, M.L.; Li, Z.; Mai, J.; Ferrari, M.; Pompa, P.P.; Shen, H. Lipopolyplex potentiates anti-tumor immunity of mRNA-based vaccination. Biomaterials 2017, 125. [Google Scholar] [CrossRef] [Green Version]
- Reichmuth, A.M.; Oberli, M.A.; Jaklenec, A.; Langer, R.; Blankschtein, D. mRNA vaccine delivery using lipid nanoparticles. Ther. Deliv. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28. [Google Scholar] [CrossRef] [PubMed]
- Hassett, K.J.; Benenato, K.E.; Jacquinet, E.; Lee, A.; Woods, A.; Yuzhakov, O.; Himansu, S.; Deterling, J.; Geilich, B.M.; Ketova, T.; et al. Optimization of Lipid Nanoparticles for Intramuscular Administration of mRNA Vaccines. Mol. Ther. Nucleic Acids 2019, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lutz, J.; Lazzaro, S.; Habbeddine, M.; Schmidt, K.E.; Baumhof, P.; Mui, B.L.; Tam, Y.K.; Madden, T.D.; Hope, M.J.; Heidenreich, R.; et al. Unmodified mRNA in LNPs constitutes a competitive technology for prophylactic vaccines. NPJ Vaccines 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the Potency of siRNA Lipid Nanoparticles for Hepatic Gene Silencing In Vivo. Angew. Chem. Int. Ed. 2012, 51. [Google Scholar] [CrossRef] [PubMed]
- Pardi, N.; LaBranche, C.C.; Ferrari, G.; Cain, D.W.; Tombácz, I.; Parks, R.J.; Muramatsu, H.; Mui, B.L.; Tam, Y.K.; Karikó, K.; et al. Characterization of HIV-1 Nucleoside-Modified mRNA Vaccines in Rabbits and Rhesus Macaques. Mol. Ther. Nucleic Acids 2019, 15. [Google Scholar] [CrossRef] [Green Version]
- Guevara, M.L.; Jilesen, Z.; Stojdl, D.; Persano, S. Codelivery of mRNA with α-Galactosylceramide Using a New Lipopolyplex Formulation Induces a Strong Antitumor Response upon Intravenous Administration. ACS Omega 2019, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.; Zheng, T.; Li, M.; Zhong, X.; Tang, Y.; Qin, M.; Sun, X. Optimization of an mRNA vaccine assisted with cyclodextrin-polyethyleneimine conjugates. Drug Deliv. Transl. Res. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Mai, Y.; Guo, J.; Zhao, Y.; Ma, S.; Hou, Y.; Yang, J. Intranasal delivery of cationic liposome-protamine complex mRNA vaccine elicits effective anti-tumor immunity. Cell. Immunol. 2020, 354. [Google Scholar] [CrossRef] [PubMed]
- Moyo, N.; Vogel, A.B.; Buus, S.; Erbar, S.; Wee, E.G.; Sahin, U.; Hanke, T. Efficient Induction of T Cells against Conserved HIV-1 Regions by Mosaic Vaccines Delivered as Self-Amplifying mRNA. Mol. Ther. Methods Clin. Dev. 2019, 12. [Google Scholar] [CrossRef] [Green Version]
- Leal, L.; Lucero, C.; Gatell, J.M.; Gallart, T.; Plana, M.; García, F. New challenges in therapeutic vaccines against HIV infection. Expert Rev. Vaccines 2017, 16, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Dybul, M.; Attoye, T.; Baptiste, S.; Cherutich, P.; Dabis, F.; Deeks, S.G.; Dieffenbach, C.; Doehle, B.; Goodenow, M.M.; Jiang, A.; et al. The case for an HIV cure and how to get there. Lancet HIV 2021, 8, e51–e58. [Google Scholar] [CrossRef]
- Angel, J.B.; Routy, J.P.; Tremblay, C.; Ayers, D.; Woods, R.; Singer, J.; Bernard, N.; Kovacs, C.; Smaill, F.; Gurunathan, S.; et al. A randomized controlled trial of HIV therapeutic vaccination using ALVAC with or without Remune. AIDS 2011, 25, 731–739. [Google Scholar] [CrossRef]
- Gorse, G.J.; Simionescu, R.E.; Patel, G.B. Cellular immune responses in asymptomatic human immunodeficiency virus type 1 (HIV-1) infection and effects of vaccination with recombinant envelope glycoprotein of HIV-1. Clin. Vaccine Immunol. 2006, 13, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Dinges, W.; Girard, P.M.; Podzamczer, D.; Brockmeyer, N.H.; García, F.; Harrer, T.; Lelievre, J.D.; Frank, I.; De Verdière, N.C.; Yeni, G.P.; et al. The F4/AS01B HIV-1 Vaccine Candidate Is Safe and Immunogenic, but Does Not Show Viral Efficacy in Antiretroviral Therapy-Naive, HIV-1-Infected Adults: A Randomized Controlled Trial. Medicine 2016, 95. [Google Scholar] [CrossRef]
- Dorrell, L.; Yang, H.; Iversen, A.K.N.; Conlon, C.; Suttill, A.; Lancaster, M.; Dong, T.; Cebere, I.; Edwards, A.; Rowland-Jones, S.; et al. Therapeutic immunization of highly active antiretroviral therapy-treated HIV-1-infected patients: Safety and immunogenicity of an HIV-1 gag/poly-epitope DNA vaccine. AIDS 2005, 19, 1321–1323. [Google Scholar] [CrossRef]
- García, F.; Climent, N.; Guardo, A.C.; Gil, C.; León, A.; Autran, B.; Lifson, J.D.; Martínez-Picado, J.; Dalmau, J.; Clotet, B.; et al. A dendritic cell-based vaccine elicits T cell responses associated with control of HIV-1 replication. Sci. Transl. Med. 2013, 5, 166ra2. [Google Scholar] [CrossRef]
- Papagno, L.; Alter, G.; Assoumou, L.; Murphy, R.L.; Garcia, F.; Clotet, B.; Larsen, M.; Braibant, M.; Marcelin, A.G.; Costagliola, D.; et al. Comprehensive analysis of virus-specific T-cells provides clues for the failure of therapeutic immunization with ALVAC-HIV vaccine. AIDS 2011, 25, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacobson, J.M.; Routy, J.P.; Welles, S.; DeBenedette, M.; Tcherepanova, I.; Angel, J.B.; Asmuth, D.M.; Stein, D.K.; Baril, J.G.; McKellar, M.; et al. Dendritic cell immunotherapy for HIV-1 infection using autologous HIV-1 RNA: A randomized, double-blind, placebo-controlled clinical trial. J. Acquir. Immune Defic. Syndr. 2016, 72, 31. [Google Scholar] [CrossRef] [Green Version]
- Gay, C.L.; Debenedette, M.A.; Tcherepanova, I.Y.; Gamble, A.; Lewis, W.E.; Cope, A.B.; Kuruc, J.D.; McGee, K.S.; Kearney, M.F.; Coffin, J.M.; et al. Immunogenicity of AGS-004 Dendritic Cell Therapy in Patients Treated during Acute HIV Infection. AIDS Res. Hum. Retrovir. 2018, 34, 111–122. [Google Scholar] [CrossRef]
- Gay, C.L.; Kuruc, J.D.; Falcinelli, S.D.; Warren, J.A.; Reifeis, S.A.; Kirchherr, J.L.; James, K.S.; Dewey, M.G.; Helms, A.; Allard, B.; et al. Assessing the impact of AGS-004, a dendritic cell-based immunotherapy, and vorinostat on persistent HIV-1 Infection. Sci. Rep. 2020, 10, 5134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gandhi, R.T.; Kwon, D.S.; Macklin, E.A.; Shopis, J.R.; McLean, A.P.; McBrine, N.; Flynn, T.; Peter, L.; Sbrolla, A.; Kaufmann, D.E.; et al. Immunization of HIV-1-infected persons with autologous dendritic cells transfected with mRNA encoding HIV-1 Gag and Nef: Results of a randomized, placebo-controlled clinical trial. J. Acquir. Immune Defic. Syndr. 2016, 71, 246. [Google Scholar] [CrossRef] [Green Version]
- de Jong, W.; Leal, L.; Buyze, J.; Pannus, P.; Guardo, A.; Salgado, M.; Mothe, B.; Molto, J.; Moron-Lopez, S.; Gálvez, C.; et al. Therapeutic vaccine in chronically Hiv-1-infected patients: A randomized, double-blind, placebocontrolled phase IIa trial with HTI-Trimix. Vaccines 2019, 9, 209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leal, L.; Guardo, A.C.; Morón-López, S.; Salgado, M.; Mothe, B.; Heirman, C.; Pannus, P.; Vanham, G.; van den Ham, H.J.; Gruters, R.; et al. Phase I clinical trial of an intranodally administered mRNA-based therapeutic vaccine against HIV-1 infection. AIDS 2018, 32, 2533–2545. [Google Scholar] [CrossRef] [Green Version]
- Erratum: Preclinical evaluation of an mRNA HIV vaccine combining rationally selected antigenic sequences and adjuvant signals (HTI-TriMix): Erratum Phase I clinical trial of an intranodally administered mRNA-based therapeutic vaccine against HIV-1 infecti. AIDS 2019. [CrossRef]
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-Based Vaccines. Pharmaceutics 2020, 12, 102. [Google Scholar] [CrossRef] [Green Version]
- Pardi, N.; Weissman, D. Nucleoside Modified mRNA Vaccines for Infectious Diseases. In RNA Vaccines; Humana Press: New York, NY, USA, 2017; pp. 109–121. [Google Scholar]
- Pardi, N.; Hogan, M.J.; Naradikian, M.S.; Parkhouse, K.; Cain, D.W.; Jones, L.; Moody, M.A.; Verkerke, H.P.; Myles, A.; Willis, E.; et al. Nucleoside-modified mRNA vaccines induce potent T follicular helper and germinal center B cell responses. J. Exp. Med. 2018, 215, 1571–1588. [Google Scholar] [CrossRef]
- Xu, S.; Yang, K.; Li, R.; Zhang, L. mRNA Vaccine Era—Mechanisms, Drug Platform and Clinical Prospection. Int. J. Mol. Sci. 2020, 21, 6582. [Google Scholar] [CrossRef]
- Edwards, D.K.; Jasny, E.; Yoon, H.; Horscroft, N.; Schanen, B.; Geter, T.; Fotin-Mleczek, M.; Petsch, B.; Wittman, V. Adjuvant effects of a sequence-engineered mRNA vaccine: Translational profiling demonstrates similar human and murine innate response. J. Transl. Med. 2017, 15, 1. [Google Scholar] [CrossRef] [Green Version]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2020, NEJMoa2035389. [Google Scholar] [CrossRef]
- Cohn, L.B.; Chomont, N.; Deeks, S.G. The Biology of the HIV-1 Latent Reservoir and Implications for Cure Strategies. Cell Host Microbe 2020, 27, 519–530. [Google Scholar] [CrossRef]
- Deeks, S.G.; Lewin, S.R.; Ross, A.L.; Ananworanich, J.; Benkirane, M.; Cannon, P.; Chomont, N.; Douek, D.; Lifson, J.D.; Lo, Y.-R.; et al. International AIDS Society global scientific strategy: Towards an HIV cure 2016. Nat. Med. 2016, 22, 839–850. [Google Scholar] [CrossRef] [PubMed]
- Bailon, L.; Mothe, B.; Berman, L.; Brander, C. Novel Approaches Towards a Functional Cure of HIV/AIDS. Drugs 2020, 80, 859–868. [Google Scholar] [CrossRef]
- Leong, Y.A.; Chen, Y.; Ong, H.S.; Wu, D.; Man, K.; Deleage, C.; Minnich, M.; Meckiff, B.J.; Wei, Y.; Hou, Z.; et al. CXCR5+ follicular cytotoxic T cells control viral infection in B cell follicles. Nat. Immunol. 2016, 17, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Pen, J.J.; Keersmaecker, B.D.; Heirman, C.; Corthals, J.; Liechtenstein, T.; Escors, D.; Thielemans, K.; Breckpot, K. Interference with PD-L1/PD-1 co-stimulation during antigen presentation enhances the multifunctionality of antigen-specific T cells. Gene Ther. 2014, 21, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Scheid, J.F.; Horwitz, J.A.; Bar-On, Y.; Kreider, E.F.; Lu, C.-L.; Lorenzi, J.C.C.; Feldmann, A.; Braunschweig, M.; Nogueira, L.; Oliveira, T.; et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature 2016, 535, 556–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McMahon, D.K.; Zheng, L.; Cyktor, J.C.; Aga, E.; Macatangay, B.J.; Godfrey, C.; Para, M.; Mitsuyasu, R.T.; Hesselgesser, J.; Dragavon, J.; et al. A phase I/II randomized, placebo-controlled trial of romidepsin in persons with HIV-1 on suppressive antiretroviral therapy to assess safety and activation of HIV-1 expression (A5315). J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]



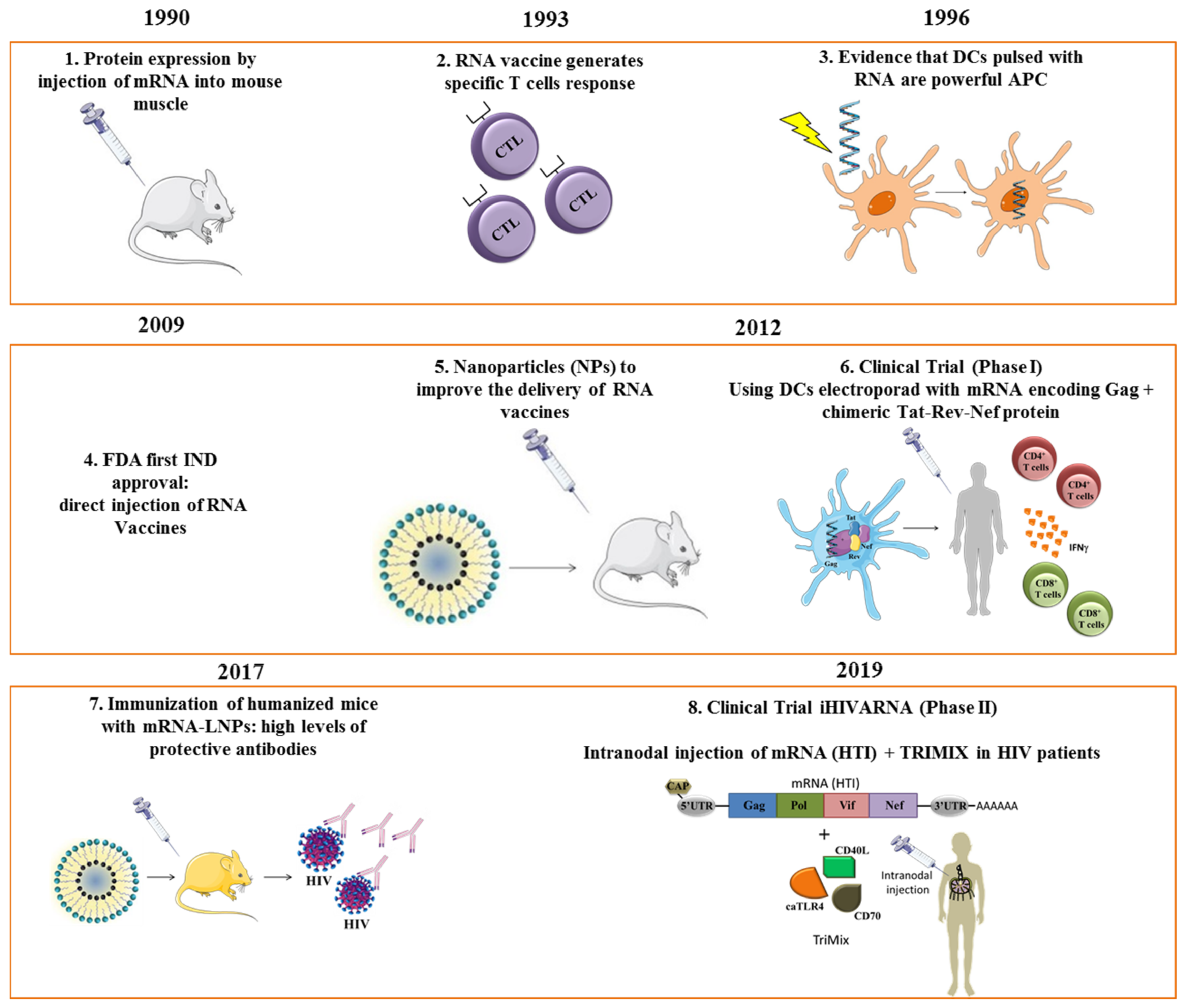{kind=link}
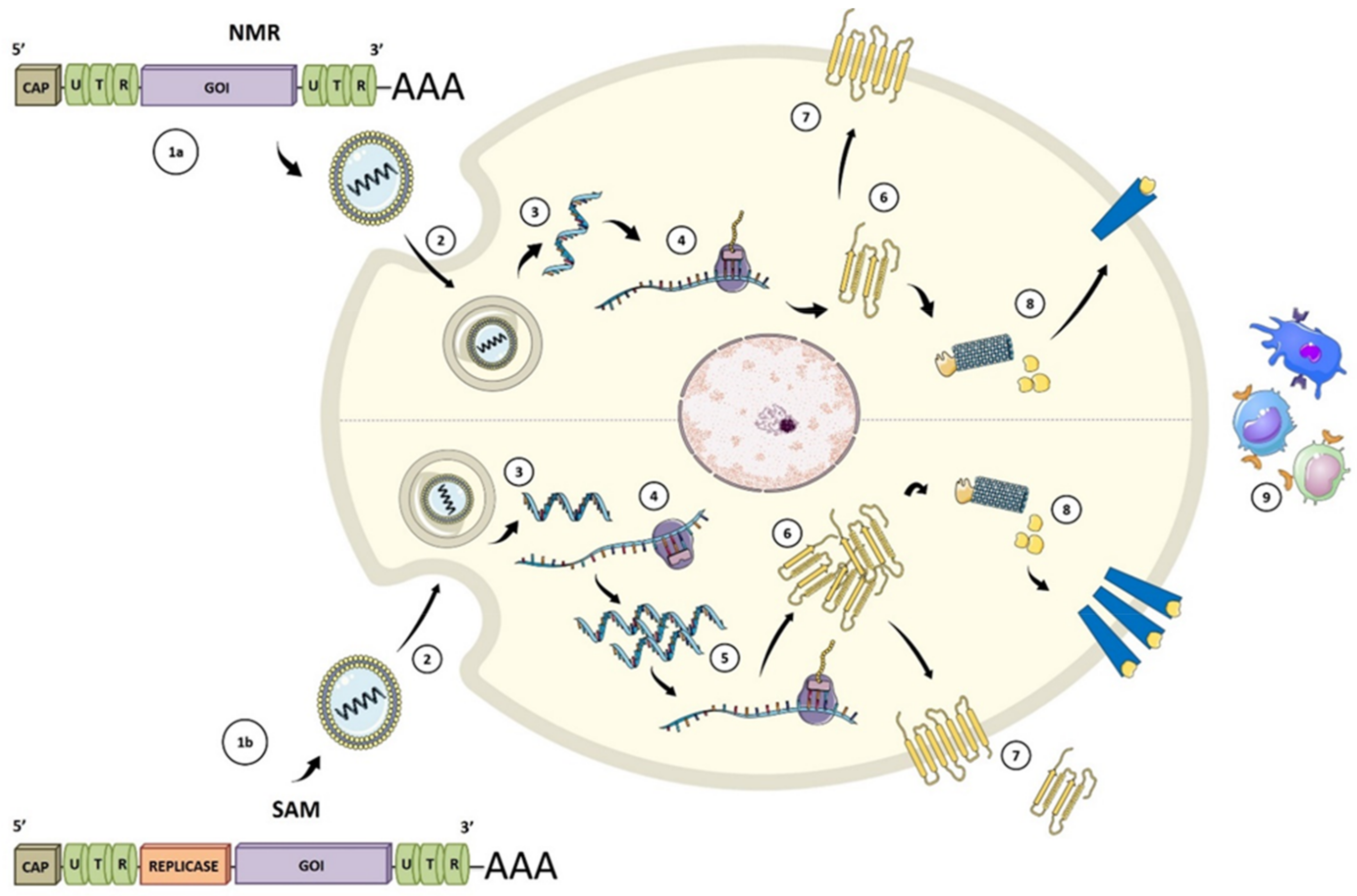{kind=link}
| Vaccines | Advantages | Disadvantages |
|---|---|---|
| RNA | Non-infectious Non-integrating Cell free Rapid and scalable production | Instability |
| Vaccine Description | Design | Main Findings |
|---|---|---|
| AGS-004 Personalized immunotherapy using electroporation of DCs with autologous amplified HIV RNAs encoding Gag, Vpr, Rev, and Nef |
|
|
| Autologous DCs electroporated with mRNA encoding Gag and a chimeric Tat, Rev, and Nef protein | Phase I/II study—ID and SC injections every 4 weeks 4 occasions [18] |
|
| Autologous DCs electroporated with mRNA encoding Gag and Nef | Randomized 2:1—4 ID injections at weeks 0, 2, 6, and 10, also received a contralateral ID injection of autologous DCs pulsed with KLH, a neo-antigen at weeks 0 and 2 [79] |
|
| iHIVARNA naked mRNA-based vaccine encoding activation signals (TriMix: CD40L + CD70 + caTLR4) combined with rationally selected antigenic sequences [HIVACAT T-cell immunogen (HTI)] sequence comprises 16 joined fragments from Gag, Pol, Vif, and Nef) |
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Esteban, I.; Pastor-Quiñones, C.; Usero, L.; Plana, M.; García, F.; Leal, L. In the Era of mRNA Vaccines, Is There Any Hope for HIV Functional Cure? Viruses 2021, 13, 501. https://doi.org/10.3390/v13030501
Esteban I, Pastor-Quiñones C, Usero L, Plana M, García F, Leal L. In the Era of mRNA Vaccines, Is There Any Hope for HIV Functional Cure? Viruses. 2021; 13(3):501. https://doi.org/10.3390/v13030501
Chicago/Turabian StyleEsteban, Ignasi, Carmen Pastor-Quiñones, Lorena Usero, Montserrat Plana, Felipe García, and Lorna Leal. 2021. "In the Era of mRNA Vaccines, Is There Any Hope for HIV Functional Cure?" Viruses 13, no. 3: 501. https://doi.org/10.3390/v13030501
APA StyleEsteban, I., Pastor-Quiñones, C., Usero, L., Plana, M., García, F., & Leal, L. (2021). In the Era of mRNA Vaccines, Is There Any Hope for HIV Functional Cure? Viruses, 13(3), 501. https://doi.org/10.3390/v13030501