
Early-Transmitted Variants and Their Evolution in a HIV-1 Positive Couple: NGS and Phylogenetic Analyses


 ,
,  ,
, 
 and
and 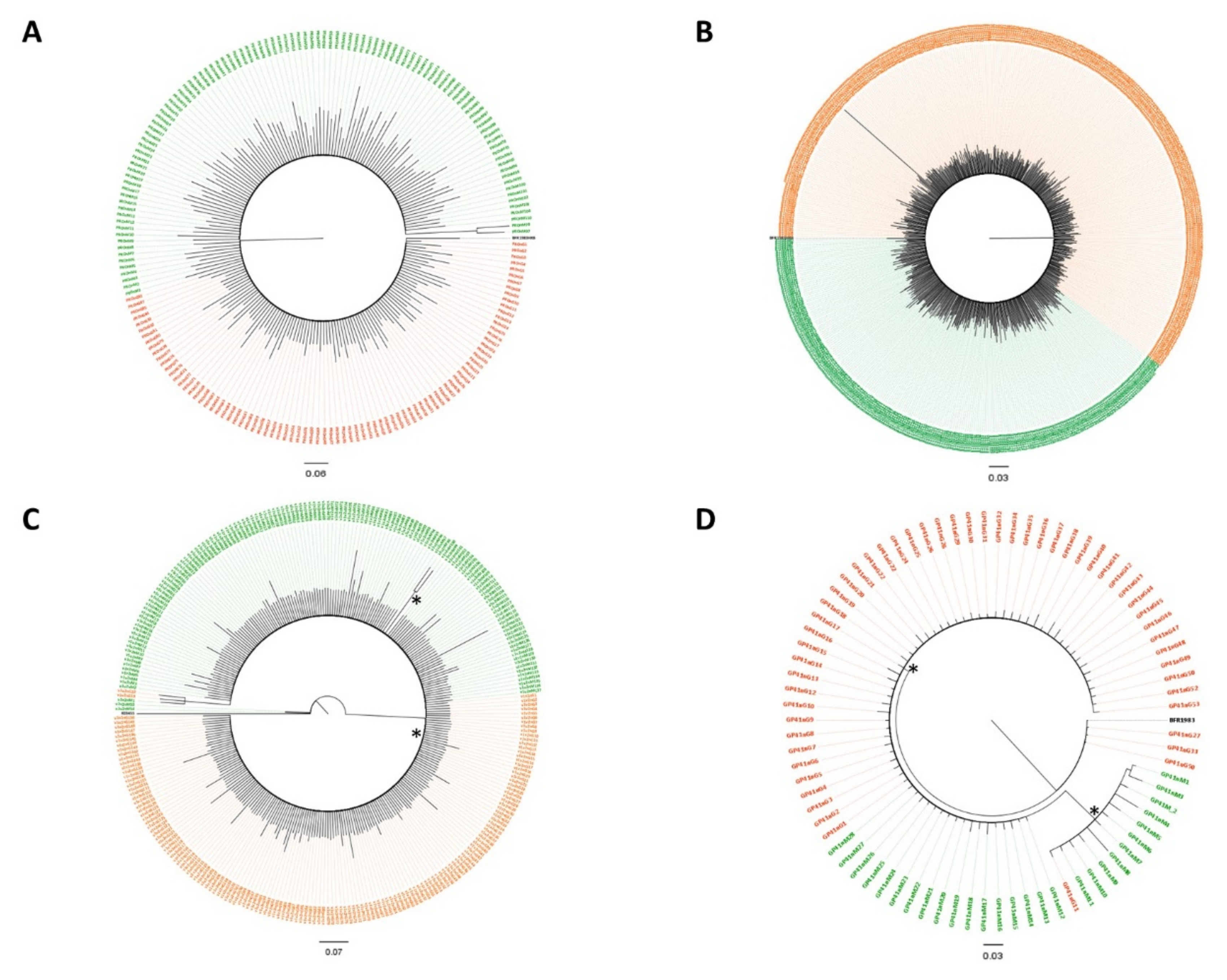{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients’ Description
2.2. DNA Extraction and HIV-1 Amplification
2.3. Next-Generation Sequencing and Data Analysis
2.4. Subtyping of Protease and RT Sequences
2.5. Phylogenetic Datasets
2.6. Phylogenetic and Phylodynamic Analyses
3. Results
3.1. Clinical Profile
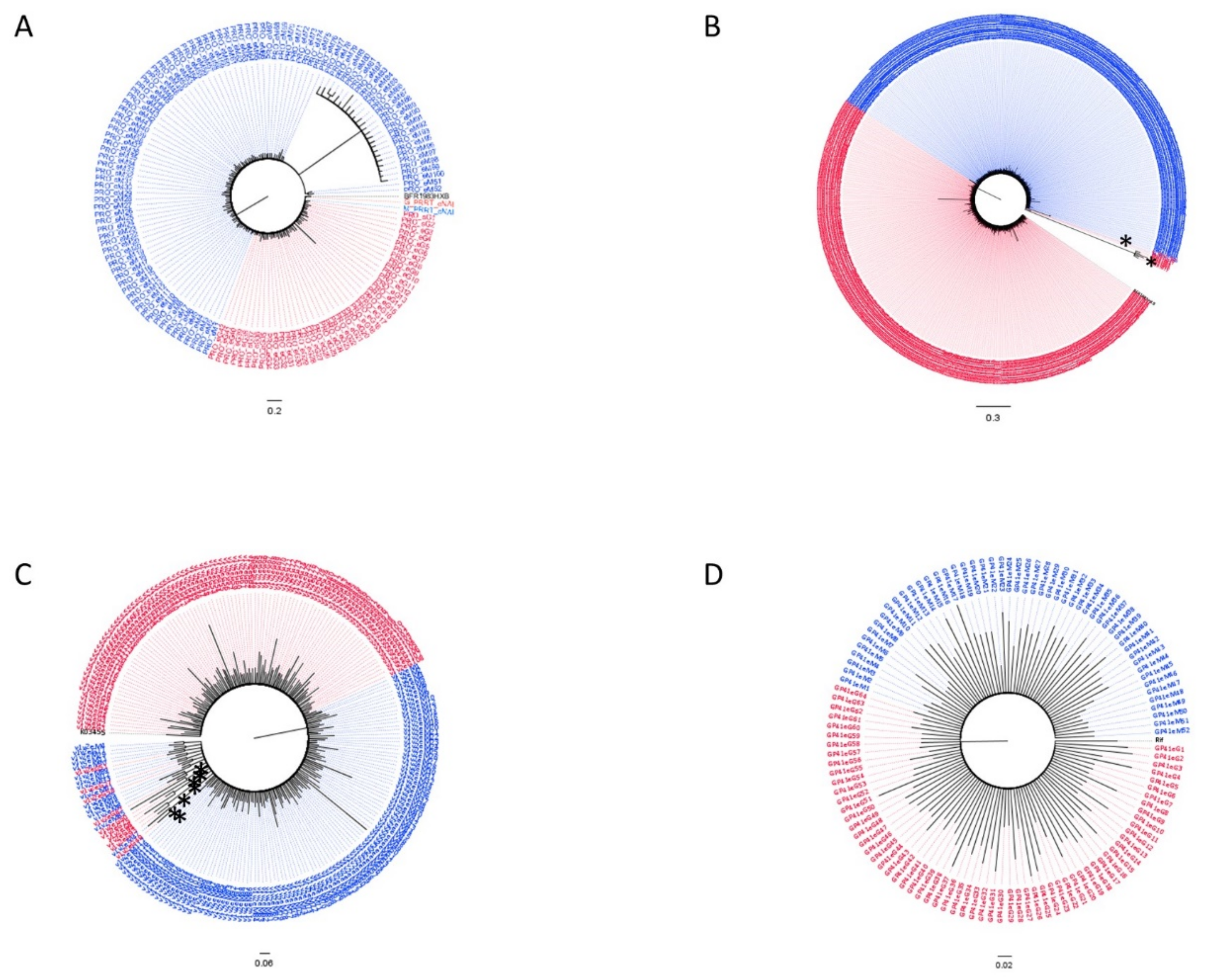
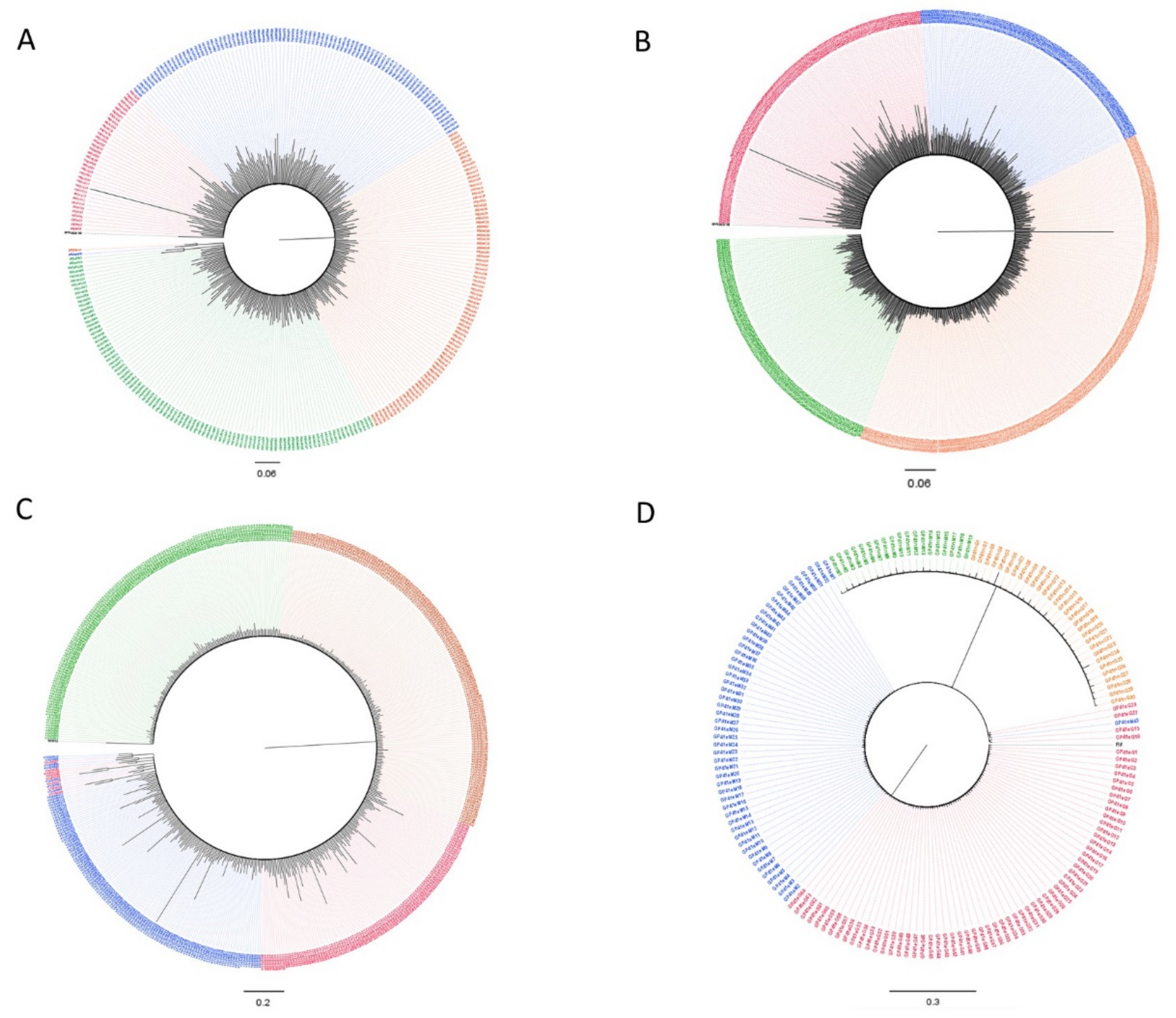
3.2. Subtypes and Genetic Distances
3.3. Variant Detection
3.4. Dated Tree Reconstruction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Perelson, A.S. Modelling Viral and Immune System Dynamics. Nat. Rev. Immunol. 2002, 2, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Lauring, A.S.; Frydman, J.; Andino, R. The Role of Mutational Robustness in RNA Virus Evolution. Nat. Rev. Microbiol. 2013, 11, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Biasin, M.; Piacentini, L.; Lo Caputo, S.; Kanari, Y.; Magri, G.; Trabattoni, D.; Naddeo, V.; Lopalco, L.; Clivio, A.; Cesana, E.; et al. Apolipoprotein B MRNA—Editing Enzyme, Catalytic Polypeptide—Like 3G: A Possible Role in the Resistance to HIV of HIV-Exposed Seronegative Individuals. J. Infect. Dis. 2007, 195, 960–964. [Google Scholar] [CrossRef]
- Cuevas, J.M.; Geller, R.; Garijo, R.; López-Aldeguer, J.; Sanjuán, R. Extremely High Mutation Rate of HIV-1 In Vivo. PLoS Biol. 2015, 13, e1002251. [Google Scholar] [CrossRef] [Green Version]
- Monajemi, M.; Woodworth, C.F.; Zipperlen, K.; Gallant, M.; Grant, M.D.; Larijani, M. Positioning of APOBEC3G/F Mutational Hotspots in the Human Immunodeficiency Virus Genome Favors Reduced Recognition by CD8+ T Cells. PLoS ONE 2014, 9, e93428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadler, H.A.; Stenglein, M.D.; Harris, R.S.; Mansky, L.M. APOBEC3G Contributes to HIV-1 Variation through Sublethal Mutagenesis. J. Virol. 2010, 84, 7396–7404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, S.B.; Swanstrom, R.; Kashuba, A.D.M.; Cohen, M.S. Bottlenecks in HIV-1 Transmission: Insights from the Study of Founder Viruses. Nat. Rev. Microbiol. 2015, 13, 414–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keele, B.F.; Giorgi, E.E.; Salazar-Gonzalez, J.F.; Decker, J.M.; Pham, K.T.; Salazar, M.G.; Sun, C.; Grayson, T.; Wang, S.; Li, H.; et al. Identification and Characterization of Transmitted and Early Founder Virus Envelopes in Primary HIV-1 Infection. Proc. Natl. Acad. Sci. USA 2008, 105, 7552–7557. [Google Scholar] [CrossRef] [Green Version]
- Abrahams, M.-R.; Anderson, J.A.; Giorgi, E.E.; Seoighe, C.; Mlisana, K.; Ping, L.-H.; Athreya, G.S.; Treurnicht, F.K.; Keele, B.F.; Wood, N.; et al. Quantitating the Multiplicity of Infection with Human Immunodeficiency Virus Type 1 Subtype C Reveals a Non-Poisson Distribution of Transmitted Variants. J. Virol. 2009, 83, 3556–3567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, G.S.; Heath, L.; Nickle, D.C.; Wong, K.G.; Leach, S.E.; Jacobs, B.; Gezahegne, S.; van ’t Wout, A.B.; Jacobson, L.P.; Margolick, J.B.; et al. HIV-1 Variation before Seroconversion in Men Who Have Sex with Men: Analysis of Acute/Early HIV Infection in the Multicenter AIDS Cohort Study. J. Infect. Dis. 2008, 197, 1011–1015. [Google Scholar] [CrossRef]
- Herbeck, J.T.; Rolland, M.; Liu, Y.; McLaughlin, S.; McNevin, J.; Zhao, H.; Wong, K.; Stoddard, J.N.; Raugi, D.; Sorensen, S.; et al. Demographic Processes Affect HIV-1 Evolution in Primary Infection before the Onset of Selective Processes. J. Virol. 2011, 85, 7523–7534. [Google Scholar] [CrossRef] [Green Version]
- Rolland, M.; Tovanabutra, S.; deCamp, A.C.; Frahm, N.; Gilbert, P.B.; Sanders-Buell, E.; Heath, L.; Magaret, C.A.; Bose, M.; Bradfield, A.; et al. Genetic Impact of Vaccination on Breakthrough HIV-1 Sequences from the STEP Trial. Nat. Med. 2011, 17, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Boeras, D.I.; Hraber, P.T.; Hurlston, M.; Evans-Strickfaden, T.; Bhattacharya, T.; Giorgi, E.E.; Mulenga, J.; Karita, E.; Korber, B.T.; Allen, S.; et al. Role of Donor Genital Tract HIV-1 Diversity in the Transmission Bottleneck. Proc. Natl. Acad. Sci. USA 2011, 108, E1156–E1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, M.; Learn, G.; Genowati, I.; McKernan, J.; Hitti, J.; Lockhart, D.; Tapia, K.; Holte, S.; Dragavon, J.; Coombs, R.; et al. Compartmentalization of HIV-1 within the Female Genital Tract Is Due to Monotypic and Low-Diversity Variants Not Distinct Viral Populations. PLoS ONE 2009, 4, e7122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diem, K.; Nickle, D.C.; Motoshige, A.; Fox, A.; Ross, S.; Mullins, J.I.; Corey, L.; Coombs, R.W.; Krieger, J.N. Male Genital Tract Compartmentalization of Human Immunodeficiency Virus Type 1 (HIV). AIDS Res. Hum. Retrovir. 2008, 24, 561–571. [Google Scholar] [CrossRef]
- Chaillon, A.; Gianella, S.; Little, S.J.; Caballero, G.; Barin, F.; Kosakovsky Pond, S.; Richman, D.D.; Smith, D.M.; Mehta, S.R. Characterizing the Multiplicity of HIV Founder Variants during Sexual Transmission among MSM. Virus Evol. 2016, 2, vew012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janes, H.; Herbeck, J.T.; Tovanabutra, S.; Thomas, R.; Frahm, N.; Duerr, A.; Hural, J.; Corey, L.; Self, S.G.; Buchbinder, S.P.; et al. HIV-1 Infections with Multiple Founders Are Associated with Higher Viral Loads than Infections with Single Founders. Nat. Med. 2015, 21, 1139–1141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, R.N.; Wymant, C.; Spriggs, R.A.; Raghwani, J.; Fraser, C.; Lythgoe, K.A. Link between the Numbers of Particles and Variants Founding New HIV-1 Infections Depends on the Timing of Transmission. Virus Evol. 2019, 5, vey038. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.-C.; Laird, G.M.; Siliciano, R.F. Measuring Reversal of HIV-1 Latency Ex Vivo Using Cells from Infected Individuals. Proc. Natl. Acad. Sci. USA 2014, 111, 6860–6861. [Google Scholar] [CrossRef] [Green Version]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-Term Follow-up Studies Confirm the Stability of the Latent Reservoir for HIV-1 in Resting CD4+ T Cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Rothenberger, M.K.; Keele, B.F.; Wietgrefe, S.W.; Fletcher, C.V.; Beilman, G.J.; Chipman, J.G.; Khoruts, A.; Estes, J.D.; Anderson, J.; Callisto, S.P.; et al. Large Number of Rebounding/Founder HIV Variants Emerge from Multifocal Infection in Lymphatic Tissues after Treatment Interruption. Proc. Natl. Acad. Sci. USA 2015, 112, E1126–E1134. [Google Scholar] [CrossRef] [Green Version]
- Maldarelli, F.; Wu, X.; Su, L.; Simonetti, F.R.; Shao, W.; Hill, S.; Spindler, J.; Ferris, A.L.; Mellors, J.W.; Kearney, M.F.; et al. Specific HIV Integration Sites Are Linked to Clonal Expansion and Persistence of Infected Cells. Science 2014, 345, 179–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonetti, F.R.; Sobolewski, M.D.; Fyne, E.; Shao, W.; Spindler, J.; Hattori, J.; Anderson, E.M.; Watters, S.A.; Hill, S.; Wu, X.; et al. Clonally Expanded CD4+ T Cells Can Produce Infectious HIV-1 in Vivo. Proc. Natl. Acad. Sci. USA 2016, 113, 1883–1888. [Google Scholar] [CrossRef] [Green Version]
- Edwards, D.; Fenizia, C.; Gold, H.; de Castro-Amarante, M.F.; Buchmann, C.; Pise-Masison, C.A.; Franchini, G. Orf-I and Orf-II-Encoded Proteins in HTLV-1 Infection and Persistence. Viruses 2011, 3, 861–885. [Google Scholar] [CrossRef] [PubMed]
- Fenizia, C.; Fiocchi, M.; Jones, K.; Parks, R.W.; Ceribelli, M.; Chevalier, S.A.; Edwards, D.; Ruscetti, F.; Pise-Masison, C.A.; Franchini, G. Human T-Cell Leukemia/Lymphoma Virus Type 1 P30, but Not P12/P8, Counteracts Toll-Like Receptor 3 (TLR3) and TLR4 Signaling in Human Monocytes and Dendritic Cells. J. Virol. 2014, 88, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Bangham, C.R.M.; Cook, L.B.; Melamed, A. HTLV-1 Clonality in Adult T-Cell Leukaemia and Non-Malignant HTLV-1 Infection. Semin. Cancer Biol. 2014, 26, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachmann, N.; von Siebenthal, C.; Vongrad, V.; Turk, T.; Neumann, K.; Beerenwinkel, N.; Bogojeska, J.; Fellay, J.; Roth, V.; Kok, Y.L.; et al. Determinants of HIV-1 Reservoir Size and Long-Term Dynamics during Suppressive ART. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, R.T.; Tashima, K.T.; Smeaton, L.M.; Vu, V.; Ritz, J.; Andrade, A.; Eron, J.J.; Hogg, E.; Fichtenbaum, C.J. Long-Term Outcomes in a Large Randomized Trial of HIV-1 Salvage Therapy: 96-Week Results of AIDS Clinical Trials Group A5241 (OPTIONS). J. Infect. Dis. 2020, 221, 1407–1415. [Google Scholar] [CrossRef]
- Peluso, M.J.; Bacchetti, P.; Ritter, K.D.; Beg, S.; Lai, J.; Martin, J.N.; Hunt, P.W.; Henrich, T.J.; Siliciano, J.D.; Siliciano, R.F.; et al. Differential Decay of Intact and Defective Proviral DNA in HIV-1–Infected Individuals on Suppressive Antiretroviral Therapy. JCI Insight 2020, 5, e132997. [Google Scholar] [CrossRef] [Green Version]
- Antar, A.A.R.; Jenike, K.M.; Jang, S.; Rigau, D.N.; Reeves, D.B.; Hoh, R.; Krone, M.R.; Keruly, J.C.; Moore, R.D.; Schiffer, J.T.; et al. Longitudinal Study Reveals HIV-1–Infected CD4+ T Cell Dynamics during Long-Term Antiretroviral Therapy. J. Clin. Investig. 2020, 130, 3543–3559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richman, D.D.; Margolis, D.M.; Delaney, M.; Greene, W.C.; Hazuda, D.; Pomerantz, R.J. The Challenge of Finding a Cure for HIV Infection. Science 2009, 323, 1304–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baral, S.; Rao, A.; Sullivan, P.; Phaswana-Mafuya, N.; Diouf, D.; Millett, G.; Musyoki, H.; Geng, E.; Mishra, S. The Disconnect between Individual-Level and Population-Level HIV Prevention Benefits of Antiretroviral Treatment. Lancet HIV 2019, 6, e632–e638. [Google Scholar] [CrossRef] [Green Version]
- Bereczky, T. U=U Is a Blessing: But Only for Patients with Access to HIV Treatment: An Essay by Tamás Bereczky. BMJ 2019, 366, l5554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchbinder, S.P.; Liu, A.Y. CROI 2019: Advances in HIV Prevention and Plans to End The Epidemic. Top. Antivir. Med. 2019, 27, 18. [Google Scholar]
- Hiv, T.L. U=U Taking off in 2017. Lancet HIV 2017, 4, e475. [Google Scholar] [CrossRef] [Green Version]
- Mayer, K.H.; de Vries, H.J. HIV and Sexually Transmitted Infections: Reconciling Estranged Bedfellows in the U = U and PrEP Era. J. Int. AIDS Soc. 2019, 22, e25357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meanley, S.; Connochie, D.; Bonett, S.; Flores, D.; Bauermeister, J. Awareness and Perceived Accuracy of Undetectable = Untransmittable: A Cross-Sectional Analysis with Implications for Treatment as Prevention among Young Men Who Have Sex with Men. Sex. Transm. Dis. 2019, 46, 733–736. [Google Scholar] [CrossRef]
- Tan, R.K.J.; Lim, J.M.; Chan, J.K.W. “Not a Walking Piece of Meat with Disease”: Meanings of Becoming Undetectable among HIV-Positive Gay, Bisexual and Other Men Who Have Sex with Men in the U = U Era. AIDS Care 2019, 0, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Jones, C.; Mieczkowski, P.; Swanstrom, R. Primer ID Validates Template Sampling Depth and Greatly Reduces the Error Rate of Next-Generation Sequencing of HIV-1 Genomic RNA Populations. J. Virol. 2015, 89, 8540–8555. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Posada, D. JModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drummond, A.J.; Rambaut, A. BEAST: Bayesian Evolutionary Analysis by Sampling Trees. BMC Evol. Biol. 2007, 7, 214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, A.; Violin, M.; Ebranati, E.; Franzetti, M.; Micheli, V.; Gismondo, M.R.; Capetti, A.; Meraviglia, P.; Simonetti, F.R.; Bozzi, G.; et al. Transmission of Resistant HIV Type 1 Variants and Epidemiological Chains in Italian Newly Diagnosed Individuals. AIDS Res. Hum. Retrovir. 2012, 28, 857–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kass, R.E.; Raftery, A.E. Bayes Factors. J. Am. Stat. Assoc. 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Bar, K.J.; Wang, S.; Decker, J.M.; Chen, Y.; Sun, C.; Salazar-Gonzalez, J.F.; Salazar, M.G.; Learn, G.H.; Morgan, C.J.; et al. High Multiplicity Infection by HIV-1 in Men Who Have Sex with Men. PLoS Pathog. 2010, 6, e1000890. [Google Scholar] [CrossRef] [Green Version]
- Yue, L.; Pfafferott, K.J.; Baalwa, J.; Conrod, K.; Dong, C.C.; Chui, C.; Rong, R.; Claiborne, D.T.; Prince, J.L.; Tang, J.; et al. Transmitted Virus Fitness and Host T Cell Responses Collectively Define Divergent Infection Outcomes in Two HIV-1 Recipients. PLoS Pathog. 2015, 11, e1004565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hughes, G.J.; Fearnhill, E.; Dunn, D.; Lycett, S.J.; Rambaut, A.; Leigh Brown, A.J. Molecular Phylodynamics of the Heterosexual HIV Epidemic in the United Kingdom. PLoS Pathog. 2009, 5, e1000590. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Wen, Y.; Wang, J.; Gong, Y.; Feng, K.; Ye, R.; Jiang, Y.; Zhao, Q.; Pan, P.; Wu, H.; et al. The Transmission and Evolution of HIV-1 Quasispecies within One Couple: A Follow-up Study Based on Next-Generation Sequencing. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Giorgi, E.E.; Keele, B.F.; Gaschen, B.; Athreya, G.S.; Salazar-Gonzalez, J.F.; Pham, K.T.; Goepfert, P.A.; Kilby, J.M.; Saag, M.S.; et al. Modeling Sequence Evolution in Acute HIV-1 Infection. J. Theor. Biol. 2009, 261, 341–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Redd, A.D.; Collinson-Streng, A.N.; Chatziandreou, N.; Mullis, C.E.; Laeyendecker, O.; Martens, C.; Ricklefs, S.; Kiwanuka, N.; Nyein, P.H.; Lutalo, T.; et al. Previously Transmitted HIV-1 Strains Are Preferentially Selected during Subsequent Sexual Transmissions. J. Infect. Dis. 2012, 206, 1433–1442. [Google Scholar] [CrossRef]
- Kariuki, S.M.; Selhorst, P.; Ariën, K.K.; Dorfman, J.R. The HIV-1 Transmission Bottleneck. Retrovirology 2017, 14, 22. [Google Scholar] [CrossRef] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lai, A.; Giacomet, V.; Bergna, A.; Zuccotti, G.V.; Zehender, G.; Clerici, M.; Trabattoni, D.; Fenizia, C. Early-Transmitted Variants and Their Evolution in a HIV-1 Positive Couple: NGS and Phylogenetic Analyses. Viruses 2021, 13, 513. https://doi.org/10.3390/v13030513
Lai A, Giacomet V, Bergna A, Zuccotti GV, Zehender G, Clerici M, Trabattoni D, Fenizia C. Early-Transmitted Variants and Their Evolution in a HIV-1 Positive Couple: NGS and Phylogenetic Analyses. Viruses. 2021; 13(3):513. https://doi.org/10.3390/v13030513
Chicago/Turabian StyleLai, Alessia, Vania Giacomet, Annalisa Bergna, Gian Vincenzo Zuccotti, Gianguglielmo Zehender, Mario Clerici, Daria Trabattoni, and Claudio Fenizia. 2021. "Early-Transmitted Variants and Their Evolution in a HIV-1 Positive Couple: NGS and Phylogenetic Analyses" Viruses 13, no. 3: 513. https://doi.org/10.3390/v13030513
APA StyleLai, A., Giacomet, V., Bergna, A., Zuccotti, G. V., Zehender, G., Clerici, M., Trabattoni, D., & Fenizia, C. (2021). Early-Transmitted Variants and Their Evolution in a HIV-1 Positive Couple: NGS and Phylogenetic Analyses. Viruses, 13(3), 513. https://doi.org/10.3390/v13030513