
Nosocomial Outbreak of SARS-CoV-2 in a “Non-COVID-19” Hospital Ward: Virus Genome Sequencing as a Key Tool to Understand Cryptic Transmission

 , ,
, ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Population, Specimen Collection and RT-PCR Testing
2.2. Genome Sequencing and Bioinformatics
3. Results and Discussion
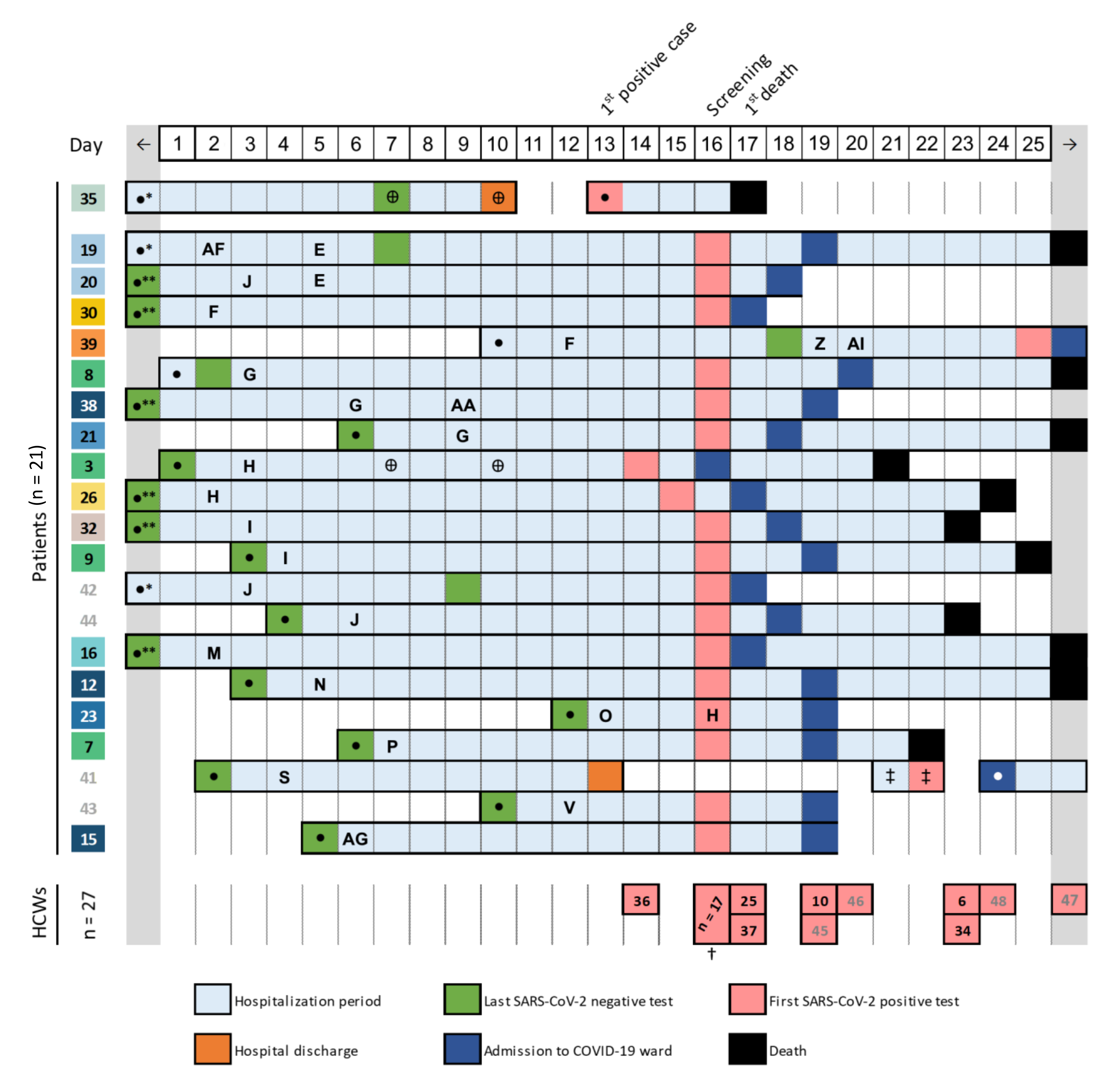
3.1. Detection of a COVID-19 Nosocomial Outbreak
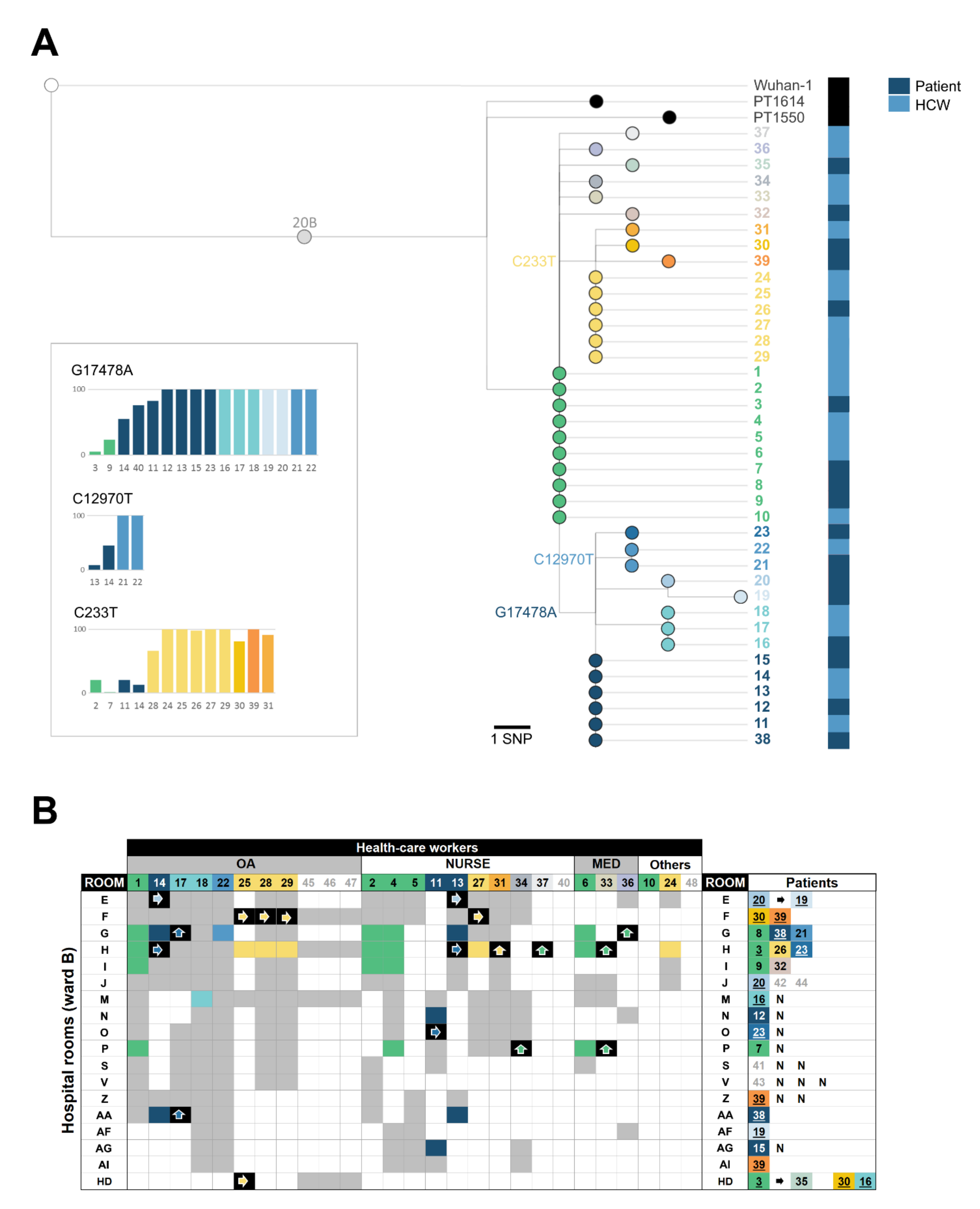
3.2. Investigation of Nosocomial Transmission of SARS-Cov-2 with In-Depth Contact Tracing and Viral Genomic Data
3.3. Genomic Characterization of the SARS-CoV-2 Outbreak Variant
3.4. Reconstructing SARS-CoV-2 Nosocomial Transmission Trajectories
3.5. Searching for the Index Case
3.6. Outbreak Follow-Up: Implemented Measures and Impact on Subsequent Outbreaks
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. Author Correction: A new coronavirus associated with human respiratory disease in China. Nat. Cell Biol. 2020, 580, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Zhou, Q.; He, Y.; Liu, L.; Ma, X.; Wei, X.; Jiang, N.; Liang, L.; Zheng, Y.; Ma, L.; et al. Nosocomial outbreak of COVID-19 pneumonia in Wuhan, China. Eur. Respir. J. 2020, 55, 2000544. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Coronavirus Disease (COVID-19) Weekly Epidemiological Update 8 December 2020. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports (accessed on 13 December 2020).
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Genet. 2021, 19, 141–154. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Infection Prevention and Control During Health Care When Coronavirus Disease (COVID-19) is Suspected or Confirmed. Geneva: World Health Organization. 2020. Available online: https://apps.who.int/iris/rest/bitstreams/1284718/retrieve (accessed on 11 October 2020).
- European Centre for Disease Prevention and Control. Infection Prevention and Control and Preparedness for COVID-19 in Healthcare Settings, 5th ed.; ECDC: Stockholm, Sweden, 2020; Available online: https://www.ecdc.europa.eu/sites/default/files/documents/Infection-prevention-and-control-in-healthcare-settings-COVID-19_5th_update.pdf (accessed on 11 October 2020).
- NBSP; The Lancet. COVID-19: Protecting Health Care Workers. 21 March 2020. Available online: https://www.thelancet.com/action/showPdf?pii=S0140-6736%2820%2930644-9 (accessed on 28 March 2020).
- Black, J.R.M.; Bailey, C.; Przewrocka, J.; Dijkstra, K.K.; Swanton, C. COVID-19: The case for health-care worker screening to prevent hospital transmission. Lancet 2020, 395, 1418–1420. [Google Scholar] [CrossRef]
- Zhan, M.; Qin, Y.; Xue, X.; Zhu, S. Death from Covid-19 of 23 Health Care Workers in China. N. Engl. J. Med. 2020, 382, 2267–2268. [Google Scholar] [CrossRef] [PubMed]
- Lucey, M.; Macori, G.; Mullane, N.; Sutton-Fitzpatrick, U.; Gonzalez, G.; Coughlan, S.; Purcell, A.; Fenelon, L.; Fanning, S.; Schaffer, K. Whole-genome Sequencing to Track Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) Transmission in Nosocomial Outbreaks. Clin. Infect. Dis. 2020, 1433. [Google Scholar] [CrossRef] [PubMed]
- Heinzerling, A.; Stuckey, M.J.; Scheuer, T.; Xu, K.; Perkins, K.M.; Resseger, H.; Magill, S.; Verani, J.R.; Jain, S.; Acosta, M.; et al. Transmission of COVID-19 to Health Care Personnel During Exposures to a Hospitalized Patient—Solano County, California, February 2020. MMWR Morb. Mortal. Wkly. Rep. 2020, 69, 472–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rickman, H.M.; Rampling, T.; Shaw, K.; Martinez-Garcia, G.; Hail, L.; Coen, P.; Shahmanesh, M.; Shin, G.Y.; Nastouli, E.; Houlihan, C.F. Nosocomial Transmission of Coronavirus Disease 2019: A Retrospective Study of 66 Hospital-acquired Cases in a London Teaching Hospital. Clin. Infect. Dis. 2021, 72, 690–693. [Google Scholar] [CrossRef] [PubMed]
- Rivett, L.; Sridhar, S.; Sparkes, D.; Routledge, M.; Jones, N.K.; Forrest, S.; Young, J.; Pereira-Dias, J.; Hamilton, W.L.; Ferris, M.; et al. Screening of healthcare workers for SARS-CoV-2 highlights the role of asymptomatic carriage in COVID-19 transmission. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Meredith, L.W.; Hamilton, W.L.; Warne, B.; Houldcroft, C.J.; Hosmillo, M.; Jahun, A.S.; Curran, M.D.; Parmar, S.; Caller, L.G.; Caddy, S.L.; et al. Rapid implementation of SARS-CoV-2 sequencing to investigate cases of health-care associated COVID-19: A prospective genomic surveillance study. Lancet Infect. Dis. 2020, 20, 1263–1271. [Google Scholar] [CrossRef]
- Safdar, N.; Moreno, G.K.; Braun, K.M.; Friedrich, T.C.; O’Connor, D.H. Using Virus Sequencing to Determine Source of SARS-CoV-2 Transmission for Healthcare Worker. Emerg. Infect. Dis. 2020, 26, 2489–2491. [Google Scholar] [CrossRef] [PubMed]
- Sikkema, R.S.; Pas, S.D.; Nieuwenhuijse, D.F.; O’Toole, Á.; Verweij, J.; Van der Linden, A.; Chestakova, I.; Schapendonk, C.; Pronk, M.; Lexmond, P.; et al. COVID-19 in health-care workers in three hospitals in the south of the Netherlands: A cross-sectional study. Lancet Infect. Dis. 2020, 20, 1273–1280. [Google Scholar] [CrossRef]
- Quick, J.; Grubaugh, N.D.; Pullan, S.T.; Claro, I.M.; Smith, A.D.; Gangavarapu, K.; Oliveira, G.; Robles-Sikisaka, R.; Rogers, T.F.; Beutler, N.A.; et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017, 12, 1261–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borges, V.; Isidro, J.; Cortes-Martins, H.; Duarte, S.; Vieira, L.; Leite, R.; Gordo, I.; Caetano, C.P.; Nunes, B.; Sá, R.; et al. Massive dissemination of a SARS-CoV-2 Spike Y839 variant in Portugal. Emerg. Microbes Infect. 2020, 9, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Borges, V.; Pinheiro, M.; Pechirra, P.; Guiomar, R.; Gomes, J.P. INSaFLU: An automated open web-based bioinformatics suite “from-reads” for influenza whole-genome-sequencing-based surveillance. Genome Med. 2018, 10, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.A.; Neher, R. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Triboli, L.; Fornasari, E.; Ray, F.; Giorgi, F.M. Coronapp: A web application to annotate and monitor SARS-CoV-2 mutations. J. Med. Virol. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Alharbi, J.; Jackson, D.; Usher, K. The potential for COVID-19 to contribute to compassion fatigue in critical care nurses. J. Clin. Nurs. 2020, 29, 2762–2764. [Google Scholar] [CrossRef] [PubMed]
- Shanafelt, T.; Ripp, J.; Trockel, M. Understanding and Addressing Sources of Anxiety Among Health Care Professionals During the COVID-19 Pandemic. JAMA 2020, 323, 2133. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Health-Care Workers (n = 27) | |
|---|---|
| Sex | |
| Male | 4 (14.3%) |
| Female | 24 (85.7%) |
| Age, years | 32 (22–54) |
| Activity | |
| Medical | 3 (11.1%) |
| Nurse | 10 (37.0%) |
| Operational assistant | 12 (44.4%) |
| Other | 2 (7.4%) |
| Symptoms * (time of testing) | |
| Yes | 3 (11.1%) |
| No | 24 (88.9%) |
| Clinical outcome | |
| Recovery | 27 (100%) |
| Death | 0 (0%) |
| Patients (n = 21) | |
| Sex | |
| Male | 10 (47.6%) |
| Female | 11 (52.4%) |
| Age, years | 82 (46–92) |
| >70 | 19 (90.5%) |
| Comorbidities | |
| Yes | 18 (85.7%) |
| No | 3 (14.3%) |
| Symptoms * (time of testing) | |
| Yes | 6 (28.6%) |
| No | 15 (71.4%) |
| Clinical outcome | |
| Recovery | 9 (42.9%) |
| Death | 12 (57.1%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Borges, V.; Isidro, J.; Macedo, F.; Neves, J.; Silva, L.; Paiva, M.; Barata, J.; Catarino, J.; Ciobanu, L.; Duarte, S.; et al. Nosocomial Outbreak of SARS-CoV-2 in a “Non-COVID-19” Hospital Ward: Virus Genome Sequencing as a Key Tool to Understand Cryptic Transmission. Viruses 2021, 13, 604. https://doi.org/10.3390/v13040604
Borges V, Isidro J, Macedo F, Neves J, Silva L, Paiva M, Barata J, Catarino J, Ciobanu L, Duarte S, et al. Nosocomial Outbreak of SARS-CoV-2 in a “Non-COVID-19” Hospital Ward: Virus Genome Sequencing as a Key Tool to Understand Cryptic Transmission. Viruses. 2021; 13(4):604. https://doi.org/10.3390/v13040604
Chicago/Turabian StyleBorges, Vítor, Joana Isidro, Filipe Macedo, José Neves, Luís Silva, Mário Paiva, José Barata, Judite Catarino, Liliana Ciobanu, Sílvia Duarte, and et al. 2021. "Nosocomial Outbreak of SARS-CoV-2 in a “Non-COVID-19” Hospital Ward: Virus Genome Sequencing as a Key Tool to Understand Cryptic Transmission" Viruses 13, no. 4: 604. https://doi.org/10.3390/v13040604
APA StyleBorges, V., Isidro, J., Macedo, F., Neves, J., Silva, L., Paiva, M., Barata, J., Catarino, J., Ciobanu, L., Duarte, S., Vieira, L., Guiomar, R., & Gomes, J. P. (2021). Nosocomial Outbreak of SARS-CoV-2 in a “Non-COVID-19” Hospital Ward: Virus Genome Sequencing as a Key Tool to Understand Cryptic Transmission. Viruses, 13(4), 604. https://doi.org/10.3390/v13040604