
Host and Viral Factors Involved in Nuclear Egress of Herpes Simplex Virus 1


Abstract
:1. Introduction
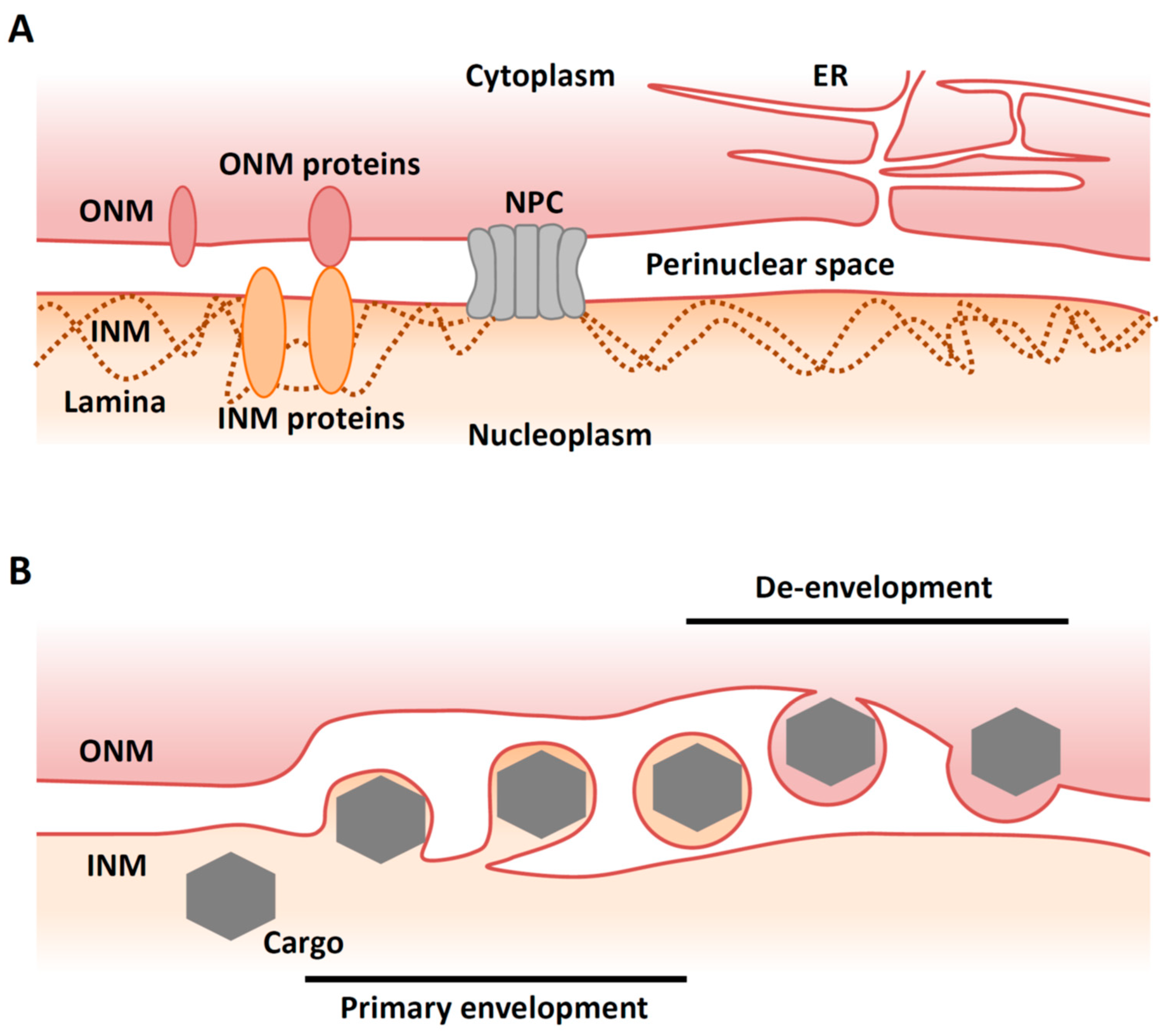
2. Overview of Nuclear Egress of Herpesviruses
3. The Nuclear Egress Complex (NEC)
4. Primary Envelopment
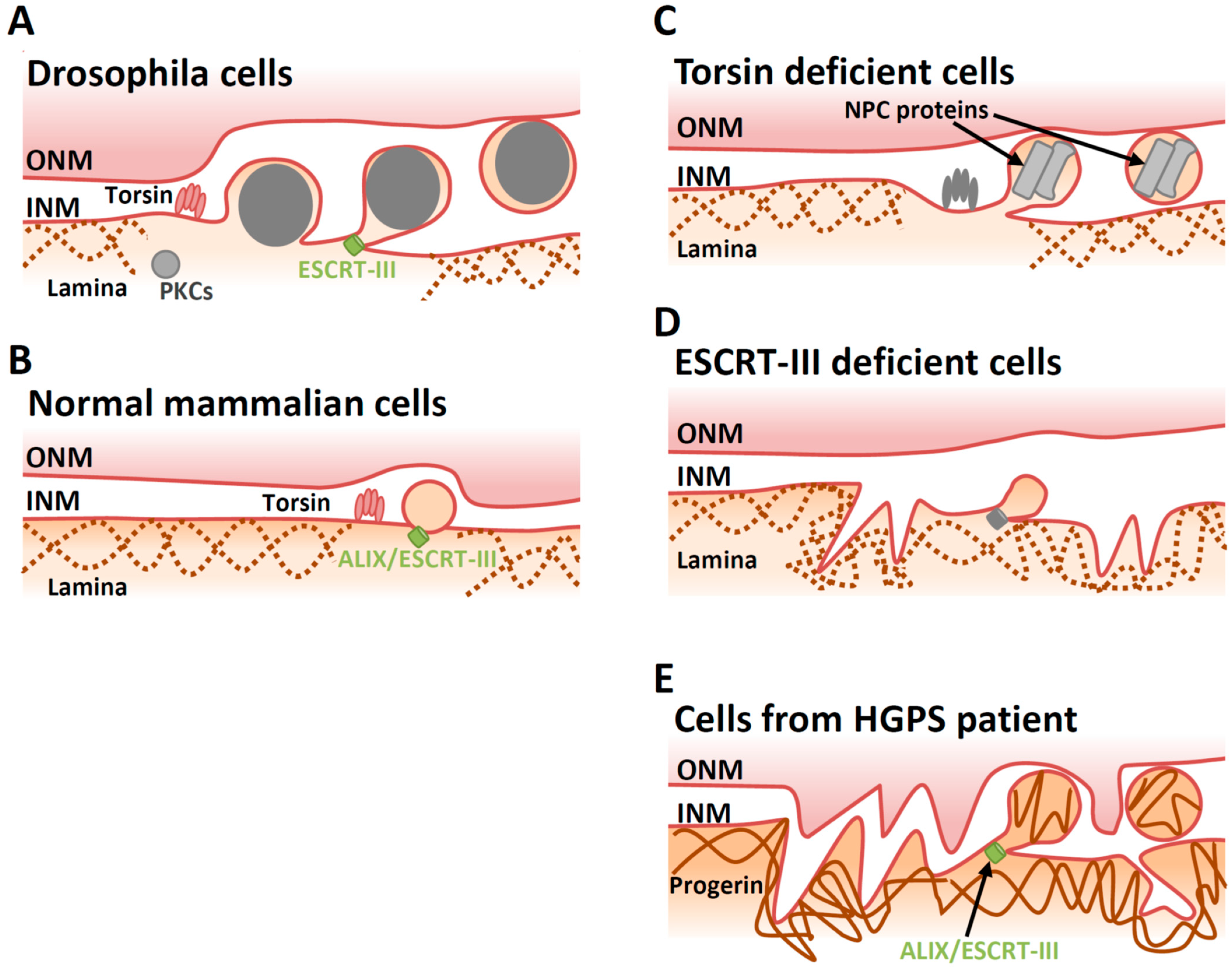
4.1. Lamina Dissociation
4.2. Deformation of the INM
4.3. Factors Involved in INM Deformation
4.4. Scission at the INM during Primary Envelopment
5. De-Envelopment
5.1. Overview of De-Envelopment
5.2. Cellular Factors Involved in De-Envelopment
6. Vesicle-Mediated Nucleocytoplasmic Transport in Uninfected Cells
7. Concluding Remarks
Funding
Conflicts of Interest
References
- Hetzer, M.W. The nuclear envelope. Cold Spring Harb. Perspect. Biol. 2010, 2, a000539. [Google Scholar] [CrossRef]
- Crisp, M.; Burke, B. The nuclear envelope as an integrator of nuclear and cytoplasmic architecture. FEBS Lett. 2008, 582, 2023–2032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paci, G.; Caria, J.; Lemke, E.A. Cargo transport through the nuclear pore complex at a glance. J. Cell Sci. 2021, 134. [Google Scholar] [CrossRef] [PubMed]
- Hagen, C.; Dent, K.C.; Zeev-Ben-Mordehai, T.; Grange, M.; Bosse, J.B.; Whittle, C.; Klupp, B.G.; Siebert, C.A.; Vasishtan, D.; Bauerlein, F.J.; et al. Structural Basis of Vesicle Formation at the Inner Nuclear Membrane. Cell 2015, 163, 1692–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Speese, S.D.; Ashley, J.; Jokhi, V.; Nunnari, J.; Barria, R.; Li, Y.; Ataman, B.; Koon, A.; Chang, Y.T.; Li, Q.; et al. Nuclear envelope budding enables large ribonucleoprotein particle export during synaptic Wnt signaling. Cell 2012, 149, 832–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellet, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Lippincott-Williams &Wilkins: Philadelphia, PA, USA, 2013; pp. 1802–1822. [Google Scholar]
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes simplex viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Lippincott-Williams &Wilkins: Philadelphia, PA, USA, 2013; pp. 1823–1897. [Google Scholar]
- Schulz, K.S.; Klupp, B.G.; Granzow, H.; Passvogel, L.; Mettenleiter, T.C. Herpesvirus nuclear egress: Pseudorabies Virus can simultaneously induce nuclear envelope breakdown and exit the nucleus via the envelopment-deenvelopment-pathway. Virus Res. 2015, 209, 76–86. [Google Scholar] [CrossRef]
- Leuzinger, H.; Ziegler, U.; Schraner, E.M.; Fraefel, C.; Glauser, D.L.; Heid, I.; Ackermann, M.; Mueller, M.; Wild, P. Herpes simplex virus 1 envelopment follows two diverse pathways. J. Virol. 2005, 79, 13047–13059. [Google Scholar] [CrossRef] [Green Version]
- Wild, P.; Senn, C.; Manera, C.L.; Sutter, E.; Schraner, E.M.; Tobler, K.; Ackermann, M.; Ziegler, U.; Lucas, M.S.; Kaech, A. Exploring the nuclear envelope of herpes simplex virus 1-infected cells by high-resolution microscopy. J. Virol. 2009, 83, 408–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettenleiter, T.C. Vesicular Nucleo-Cytoplasmic Transport-Herpesviruses as Pioneers in Cell Biology. Viruses 2016, 8, 266. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.E.; Van Sant, C.; Krug, P.W.; Sears, A.E.; Roizman, B. The null mutant of the U(L)31 gene of herpes simplex virus 1: Construction and phenotype in infected cells. J. Virol. 1997, 71, 8307–8315. [Google Scholar] [CrossRef] [Green Version]
- Roller, R.J.; Zhou, Y.; Schnetzer, R.; Ferguson, J.; DeSalvo, D. Herpes simplex virus type 1 U(L)34 gene product is required for viral envelopment. J. Virol. 2000, 74, 117–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Primary envelopment of pseudorabies virus at the nuclear membrane requires the UL34 gene product. J. Virol. 2000, 74, 10063–10073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Osterrieder, N.; Mettenleiter, T.C. The interacting UL31 and UL34 gene products of pseudorabies virus are involved in egress from the host-cell nucleus and represent components of primary enveloped but not mature virions. J. Virol. 2002, 76, 364–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farina, A.; Feederle, R.; Raffa, S.; Gonnella, R.; Santarelli, R.; Frati, L.; Angeloni, A.; Torrisi, M.R.; Faggioni, A.; Delecluse, H.J. BFRF1 of Epstein-Barr virus is essential for efficient primary viral envelopment and egress. J. Virol. 2005, 79, 3703–3712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popa, M.; Ruzsics, Z.; Lotzerich, M.; Dolken, L.; Buser, C.; Walther, P.; Koszinowski, U.H. Dominant negative mutants of the murine cytomegalovirus M53 gene block nuclear egress and inhibit capsid maturation. J. Virol. 2010, 84, 9035–9046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiba, C.; Daikoku, T.; Goshima, F.; Takakuwa, H.; Yamauchi, Y.; Koiwai, O.; Nishiyama, Y. The UL34 gene product of herpes simplex virus type 2 is a tail-anchored type II membrane protein that is significant for virus envelopment. J. Gen. Virol. 2000, 81, 2397–2405. [Google Scholar] [CrossRef]
- Reynolds, A.E.; Ryckman, B.J.; Baines, J.D.; Zhou, Y.; Liang, L.; Roller, R.J. U(L)31 and U(L)34 proteins of herpes simplex virus type 1 form a complex that accumulates at the nuclear rim and is required for envelopment of nucleocapsids. J. Virol. 2001, 75, 8803–8817. [Google Scholar] [CrossRef] [Green Version]
- Bigalke, J.M.; Heldwein, E.E. Structural basis of membrane budding by the nuclear egress complex of herpesviruses. EMBO J. 2015, 34, 2921–2936. [Google Scholar] [CrossRef]
- Zeev-Ben-Mordehai, T.; Weberruss, M.; Lorenz, M.; Cheleski, J.; Hellberg, T.; Whittle, C.; El Omari, K.; Vasishtan, D.; Dent, K.C.; Harlos, K.; et al. Crystal Structure of the Herpesvirus Nuclear Egress Complex Provides Insights into Inner Nuclear Membrane Remodeling. Cell Rep. 2015, 13, 2645–2652. [Google Scholar] [CrossRef] [Green Version]
- Lye, M.F.; Sharma, M.; El Omari, K.; Filman, D.J.; Schuermann, J.P.; Hogle, J.M.; Coen, D.M. Unexpected features and mechanism of heterodimer formation of a herpesvirus nuclear egress complex. EMBO J. 2015, 34, 2937–2952. [Google Scholar] [CrossRef] [Green Version]
- Leigh, K.E.; Sharma, M.; Mansueto, M.S.; Boeszoermenyi, A.; Filman, D.J.; Hogle, J.M.; Wagner, G.; Coen, D.M.; Arthanari, H. Structure of a herpesvirus nuclear egress complex subunit reveals an interaction groove that is essential for viral replication. Proc. Natl. Acad. Sci. USA 2015, 112, 9010–9015. [Google Scholar] [CrossRef] [Green Version]
- Walzer, S.A.; Egerer-Sieber, C.; Sticht, H.; Sevvana, M.; Hohl, K.; Milbradt, J.; Muller, Y.A.; Marschall, M. Crystal Structure of the Human Cytomegalovirus pUL50-pUL53 Core Nuclear Egress Complex Provides Insight into a Unique Assembly Scaffold for Virus-Host Protein Interactions. J. Biol. Chem. 2015, 290, 27452–27458. [Google Scholar] [CrossRef] [Green Version]
- Gruenbaum, Y.; Margalit, A.; Goldman, R.D.; Shumaker, D.K.; Wilson, K.L. The nuclear lamina comes of age. Nat. Rev. Mol. Cell Biol. 2005, 6, 21–31. [Google Scholar] [CrossRef]
- Muranyi, W.; Haas, J.; Wagner, M.; Krohne, G.; Koszinowski, U.H. Cytomegalovirus recruitment of cellular kinases to dissolve the nuclear lamina. Science 2002, 297, 854–857. [Google Scholar] [CrossRef]
- Scott, E.S.; O’Hare, P. Fate of the inner nuclear membrane protein lamin B receptor and nuclear lamins in herpes simplex virus type 1 infection. J. Virol. 2001, 75, 8818–8830. [Google Scholar] [CrossRef] [Green Version]
- Reynolds, A.E.; Liang, L.; Baines, J.D. Conformational changes in the nuclear lamina induced by herpes simplex virus type 1 require genes U(L)31 and U(L)34. J. Virol. 2004, 78, 5564–5575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson-Holley, M.; Baines, J.; Roller, R.; Knipe, D.M. Herpes simplex virus 1 U(L)31 and U(L)34 gene products promote the late maturation of viral replication compartments to the nuclear periphery. J. Virol. 2004, 78, 5591–5600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simpson-Holley, M.; Colgrove, R.C.; Nalepa, G.; Harper, J.W.; Knipe, D.M. Identification and functional evaluation of cellular and viral factors involved in the alteration of nuclear architecture during herpes simplex virus 1 infection. J. Virol. 2005, 79, 12840–12851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, R.; Baines, J.D. Herpes simplex virus type 1 infection induces activation and recruitment of protein kinase C to the nuclear membrane and increased phosphorylation of lamin B. J. Virol. 2006, 80, 494–504. [Google Scholar] [CrossRef] [Green Version]
- Bjerke, S.L.; Roller, R.J. Roles for herpes simplex virus type 1 UL34 and US3 proteins in disrupting the nuclear lamina during herpes simplex virus type 1 egress. Virology 2006, 347, 261–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, F.; Forest, T.; Baines, J.D. US3 of herpes simplex virus type 1 encodes a promiscuous protein kinase that phosphorylates and alters localization of lamin A/C in infected cells. J. Virol. 2007, 81, 6459–6470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, F.; Wills, E.G.; Park, R.; Baines, J.D. Effects of lamin A/C, lamin B1, and viral US3 kinase activity on viral infectivity, virion egress, and the targeting of herpes simplex virus U(L)34-encoded protein to the inner nuclear membrane. J. Virol. 2008, 82, 8094–8104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuny, C.V.; Chinchilla, K.; Culbertson, M.R.; Kalejta, R.F. Cyclin-dependent kinase-like function is shared by the beta- and gamma- subset of the conserved herpesvirus protein kinases. PLoS Pathog. 2010, 6, e1001092. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.P.; Huang, Y.H.; Lin, S.F.; Chang, Y.; Chang, Y.H.; Takada, K.; Chen, M.R. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 2008, 82, 11913–11926. [Google Scholar] [CrossRef] [Green Version]
- Hamirally, S.; Kamil, J.P.; Ndassa-Colday, Y.M.; Lin, A.J.; Jahng, W.J.; Baek, M.C.; Noton, S.; Silva, L.A.; Simpson-Holley, M.; Knipe, D.M.; et al. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 2009, 5, e1000275. [Google Scholar] [CrossRef] [Green Version]
- Leach, N.; Bjerke, S.L.; Christensen, D.K.; Bouchard, J.M.; Mou, F.; Park, R.; Baines, J.; Haraguchi, T.; Roller, R.J. Emerin is hyperphosphorylated and redistributed in herpes simplex virus type 1-infected cells in a manner dependent on both UL34 and US3. J. Virol. 2007, 81, 10792–10803. [Google Scholar] [CrossRef] [Green Version]
- Morris, J.B.; Hofemeister, H.; O’Hare, P. Herpes simplex virus infection induces phosphorylation and delocalization of emerin, a key inner nuclear membrane protein. J. Virol. 2007, 81, 4429–4437. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Pan, S.; Zhang, L.; Baines, J.; Roller, R.; Ames, J.; Yang, M.; Wang, J.; Chen, D.; Liu, Y.; et al. Herpes Simplex Virus 1 Induces Phosphorylation and Reorganization of Lamin A/C through the gamma134.5 Protein That Facilitates Nuclear Egress. J. Virol. 2016, 90, 10414–10422. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yang, Y.; Wu, S.; Pan, S.; Zhou, C.; Ma, Y.; Ru, Y.; Dong, S.; He, B.; Zhang, C.; et al. p32 is a novel target for viral protein ICP34.5 of herpes simplex virus type 1 and facilitates viral nuclear egress. J. Biol. Chem. 2014, 289, 35795–35805. [Google Scholar] [CrossRef] [Green Version]
- Turan, A.; Grosche, L.; Krawczyk, A.; Muhl-Zurbes, P.; Drassner, C.; Duthorn, A.; Kummer, M.; Hasenberg, M.; Voortmann, S.; Jastrow, H.; et al. Autophagic degradation of lamins facilitates the nuclear egress of herpes simplex virus type 1. J. Cell Biol. 2019, 218, 508–523. [Google Scholar] [CrossRef]
- Full, F.; van Gent, M.; Sparrer, K.M.J.; Chiang, C.; Zurenski, M.A.; Scherer, M.; Brockmeyer, N.H.; Heinzerling, L.; Sturzl, M.; Korn, K.; et al. Centrosomal protein TRIM43 restricts herpesvirus infection by regulating nuclear lamina integrity. Nat. Microbiol. 2019, 4, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, K.H.; Kennedy, B.K. When lamins go bad: Nuclear structure and disease. Cell 2013, 152, 1365–1375. [Google Scholar] [CrossRef] [Green Version]
- Silva, L.; Cliffe, A.; Chang, L.; Knipe, D.M. Role for A-type lamins in herpesviral DNA targeting and heterochromatin modulation. PLoS Pathog. 2008, 4, e1000071. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, L.; Oh, H.S.; Chang, L.; Yan, Z.; Triezenberg, S.J.; Knipe, D.M. Roles of the nuclear lamina in stable nuclear association and assembly of a herpesviral transactivator complex on viral immediate-early genes. mBio 2012, 3. [Google Scholar] [CrossRef] [Green Version]
- Oh, H.S.; Traktman, P.; Knipe, D.M. Barrier-to-Autointegration Factor 1 (BAF/BANF1) Promotes Association of the SETD1A Histone Methyltransferase with Herpes Simplex Virus Immediate-Early Gene Promoters. mBio 2015, 6, e00345-15. [Google Scholar] [CrossRef] [Green Version]
- Roller, R.J.; Bjerke, S.L.; Haugo, A.C.; Hanson, S. Analysis of a charge cluster mutation of herpes simplex virus type 1 UL34 and its extragenic suppressor suggests a novel interaction between pUL34 and pUL31 that is necessary for membrane curvature around capsids. J. Virol. 2010, 84, 3921–3934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funk, C.; Ott, M.; Raschbichler, V.; Nagel, C.H.; Binz, A.; Sodeik, B.; Bauerfeind, R.; Bailer, S.M. The Herpes Simplex Virus Protein pUL31 Escorts Nucleocapsids to Sites of Nuclear Egress, a Process Coordinated by Its N-Terminal Domain. PLoS Pathog. 2015, 11, e1004957. [Google Scholar] [CrossRef] [Green Version]
- Trus, B.L.; Newcomb, W.W.; Cheng, N.; Cardone, G.; Marekov, L.; Homa, F.L.; Brown, J.C.; Steven, A.C. Allosteric signaling and a nuclear exit strategy: Binding of UL25/UL17 heterodimers to DNA-Filled HSV-1 capsids. Mol. Cell 2007, 26, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Sheaffer, A.K.; Newcomb, W.W.; Gao, M.; Yu, D.; Weller, S.K.; Brown, J.C.; Tenney, D.J. Herpes simplex virus DNA cleavage and packaging proteins associate with the procapsid prior to its maturation. J. Virol. 2001, 75, 687–698. [Google Scholar] [CrossRef] [Green Version]
- Thurlow, J.K.; Rixon, F.J.; Murphy, M.; Targett-Adams, P.; Hughes, M.; Preston, V.G. The herpes simplex virus type 1 DNA packaging protein UL17 is a virion protein that is present in both the capsid and the tegument compartments. J. Virol. 2005, 79, 150–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newcomb, W.W.; Homa, F.L.; Brown, J.C. Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. J. Virol. 2006, 80, 6286–6294. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Baines, J.D. Selection of HSV capsids for envelopment involves interaction between capsid surface components pUL31, pUL17, and pUL25. Proc. Natl. Acad. Sci. USA 2011, 108, 14276–14281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, K.; Wills, E.; Lim, H.Y.; Zhou, Z.H.; Baines, J.D. Association of herpes simplex virus pUL31 with capsid vertices and components of the capsid vertex-specific complex. J. Virol. 2014, 88, 3815–3825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeshima, K.; Arii, J.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. Identification of the Capsid Binding Site in the Herpes Simplex Virus 1 Nuclear Egress Complex and Its Role in Viral Primary Envelopment and Replication. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Newcomb, W.W.; Fontana, J.; Winkler, D.C.; Cheng, N.; Heymann, J.B.; Steven, A.C. The Primary Enveloped Virion of Herpes Simplex Virus 1: Its Role in Nuclear Egress. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Draganova, E.B.; Zhang, J.; Zhou, Z.H.; Heldwein, E.E. Structural basis for capsid recruitment and coat formation during HSV-1 nuclear egress. Elife 2020, 9. [Google Scholar] [CrossRef]
- Klupp, B.G.; Granzow, H.; Fuchs, W.; Keil, G.M.; Finke, S.; Mettenleiter, T.C. Vesicle formation from the nuclear membrane is induced by coexpression of two conserved herpesvirus proteins. Proc. Natl. Acad. Sci. USA 2007, 104, 7241–7246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luitweiler, E.M.; Henson, B.W.; Pryce, E.N.; Patel, V.; Coombs, G.; McCaffery, J.M.; Desai, P.J. Interactions of the Kaposi’s Sarcoma-associated herpesvirus nuclear egress complex: ORF69 is a potent factor for remodeling cellular membranes. J. Virol. 2013, 87, 3915–3929. [Google Scholar] [CrossRef] [Green Version]
- Desai, P.J.; Pryce, E.N.; Henson, B.W.; Luitweiler, E.M.; Cothran, J. Reconstitution of the Kaposi’s sarcoma-associated herpesvirus nuclear egress complex and formation of nuclear membrane vesicles by coexpression of ORF67 and ORF69 gene products. J. Virol. 2012, 86, 594–598. [Google Scholar] [CrossRef] [Green Version]
- Bigalke, J.M.; Heuser, T.; Nicastro, D.; Heldwein, E.E. Membrane deformation and scission by the HSV-1 nuclear egress complex. Nat. Commun 2014, 5, 4131. [Google Scholar] [CrossRef]
- Lorenz, M.; Vollmer, B.; Unsay, J.D.; Klupp, B.G.; Garcia-Saez, A.J.; Mettenleiter, T.C.; Antonin, W. A single herpesvirus protein can mediate vesicle formation in the nuclear envelope. J. Biol. Chem. 2015, 290, 6962–6974. [Google Scholar] [CrossRef] [Green Version]
- Arii, J.; Takeshima, K.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. Roles of the Interhexamer Contact Site for Hexagonal Lattice Formation of the Herpes Simplex Virus 1 Nuclear Egress Complex in Viral Primary Envelopment and Replication. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Thaller, D.J.; Tong, D.; Marklew, C.J.; Ader, N.R.; Mannino, P.J.; Borah, S.; King, M.C.; Ciani, B.; Lusk, C.P. Direct binding of ESCRT protein Chm7 to phosphatidic acid-rich membranes at nuclear envelope herniations. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef]
- Roussel, E.; Lippe, R. Cellular Protein Kinase D Modulators Play a Role during Multiple Steps of Herpes Simplex Virus 1 Egress. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Arii, J.; Fukui, A.; Shimanaka, Y.; Kono, N.; Arai, H.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Mori, Y.; Kawaguchi, Y. Role of Phosphatidylethanolamine Biosynthesis in Herpes Simplex Virus 1-Infected Cells in Progeny Virus Morphogenesis in the Cytoplasm and in Viral Pathogenicity In Vivo. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Naldinho-Souto, R.; Browne, H.; Minson, T. Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J. Virol. 2006, 80, 2582–2584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucks, M.A.; O’Regan, K.J.; Murphy, M.A.; Wills, J.W.; Courtney, R.J. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 2007, 361, 316–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donnelly, M.; Elliott, G. Fluorescent tagging of herpes simplex virus tegument protein VP13/14 in virus infection. J. Virol. 2001, 75, 2575–2583. [Google Scholar] [CrossRef] [Green Version]
- Pomeranz, L.E.; Blaho, J.A. Modified VP22 localizes to the cell nucleus during synchronized herpes simplex virus type 1 infection. J. Virol. 1999, 73, 6769–6781. [Google Scholar] [CrossRef] [Green Version]
- Henaff, D.; Remillard-Labrosse, G.; Loret, S.; Lippe, R. Analysis of the early steps of herpes simplex virus 1 capsid tegumentation. J. Virol. 2013, 87, 4895–4906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luxton, G.W.; Lee, J.I.; Haverlock-Moyns, S.; Schober, J.M.; Smith, G.A. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 2006, 80, 201–209. [Google Scholar] [CrossRef] [Green Version]
- Leelawong, M.; Lee, J.I.; Smith, G.A. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J. Virol. 2012, 86, 6303–6314. [Google Scholar] [CrossRef] [Green Version]
- Huet, A.; Makhov, A.M.; Huffman, J.B.; Vos, M.; Homa, F.L.; Conway, J.F. Extensive subunit contacts underpin herpesvirus capsid stability and interior-to-exterior allostery. Nat. Struct. Mol. Biol. 2016, 23, 531–539. [Google Scholar] [CrossRef] [Green Version]
- Cardone, G.; Newcomb, W.W.; Cheng, N.; Wingfield, P.T.; Trus, B.L.; Brown, J.C.; Steven, A.C. The UL36 tegument protein of herpes simplex virus 1 has a composite binding site at the capsid vertices. J. Virol. 2012, 86, 4058–4064. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Kato, A.; Shindo, K.; Noda, T.; Sagara, H.; Kawaoka, Y.; Arii, J.; Kawaguchi, Y. Herpes simplex virus 1 UL47 interacts with viral nuclear egress factors UL31, UL34, and Us3 and regulates viral nuclear egress. J. Virol. 2014, 88, 4657–4667. [Google Scholar] [CrossRef] [Green Version]
- Maruzuru, Y.; Shindo, K.; Liu, Z.; Oyama, M.; Kozuka-Hata, H.; Arii, J.; Kato, A.; Kawaguchi, Y. Role of herpes simplex virus 1 immediate early protein ICP22 in viral nuclear egress. J. Virol. 2014, 88, 7445–7454. [Google Scholar] [CrossRef] [Green Version]
- Arii, J.; Watanabe, M.; Maeda, F.; Tokai-Nishizumi, N.; Chihara, T.; Miura, M.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat. Commun. 2018, 9, 3379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mou, F.; Wills, E.; Baines, J.D. Phosphorylation of the U(L)31 protein of herpes simplex virus 1 by the U(S)3-encoded kinase regulates localization of the nuclear envelopment complex and egress of nucleocapsids. J. Virol. 2009, 83, 5181–5191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano-Monreal, G.L.; Wylie, K.M.; Cao, F.; Tavis, J.E.; Morrison, L.A. Herpes simplex virus 2 UL13 protein kinase disrupts nuclear lamins. Virology 2009, 392, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynolds, A.E.; Wills, E.G.; Roller, R.J.; Ryckman, B.J.; Baines, J.D. Ultrastructural localization of the herpes simplex virus type 1 UL31, UL34, and US3 proteins suggests specific roles in primary envelopment and egress of nucleocapsids. J. Virol. 2002, 76, 8939–8952. [Google Scholar] [CrossRef] [Green Version]
- Purves, F.C.; Spector, D.; Roizman, B. The herpes simplex virus 1 protein kinase encoded by the US3 gene mediates posttranslational modification of the phosphoprotein encoded by the UL34 gene. J. Virol. 1991, 65, 5757–5764. [Google Scholar] [CrossRef] [Green Version]
- Wisner, T.W.; Wright, C.C.; Kato, A.; Kawaguchi, Y.; Mou, F.; Baines, J.D.; Roller, R.J.; Johnson, D.C. Herpesvirus gB-induced fusion between the virion envelope and outer nuclear membrane during virus egress is regulated by the viral US3 kinase. J. Virol. 2009, 83, 3115–3126. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Arii, J.; Shiratori, I.; Akashi, H.; Arase, H.; Kawaguchi, Y. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J. Virol. 2009, 83, 250–261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, A.; Yamamoto, M.; Ohno, T.; Kodaira, H.; Nishiyama, Y.; Kawaguchi, Y. Identification of proteins phosphorylated directly by the Us3 protein kinase encoded by herpes simplex virus 1. J. Virol. 2005, 79, 9325–9331. [Google Scholar] [CrossRef] [Green Version]
- Kopp, M.; Klupp, B.G.; Granzow, H.; Fuchs, W.; Mettenleiter, T.C. Identification and characterization of the pseudorabies virus tegument proteins UL46 and UL47: Role for UL47 in virion morphogenesis in the cytoplasm. J. Virol. 2002, 76, 8820–8833. [Google Scholar] [CrossRef] [Green Version]
- Fuchs, W.; Granzow, H.; Mettenleiter, T.C. A pseudorabies virus recombinant simultaneously lacking the major tegument proteins encoded by the UL46, UL47, UL48, and UL49 genes is viable in cultured cells. J. Virol. 2003, 77, 12891–12900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farnsworth, A.; Wisner, T.W.; Webb, M.; Roller, R.; Cohen, G.; Eisenberg, R.; Johnson, D.C. Herpes simplex virus glycoproteins gB and gH function in fusion between the virion envelope and the outer nuclear membrane. Proc. Natl. Acad. Sci. USA 2007, 104, 10187–10192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klupp, B.; Altenschmidt, J.; Granzow, H.; Fuchs, W.; Mettenleiter, T.C. Glycoproteins required for entry are not necessary for egress of pseudorabies virus. J. Virol. 2008, 82, 6299–6309. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, N.; Daikoku, T.; Koshizuka, T.; Yamauchi, Y.; Yoshikawa, T.; Nishiyama, Y. Subcellular localization of herpes simplex virus type 1 UL51 protein and role of palmitoylation in Golgi apparatus targeting. J. Virol. 2003, 77, 3204–3216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klupp, B.G.; Granzow, H.; Klopfleisch, R.; Fuchs, W.; Kopp, M.; Lenk, M.; Mettenleiter, T.C. Functional analysis of the pseudorabies virus UL51 protein. J. Virol. 2005, 79, 3831–3840. [Google Scholar] [CrossRef] [Green Version]
- Saiz-Ros, N.; Czapiewski, R.; Epifano, I.; Stevenson, A.; Swanson, S.K.; Dixon, C.R.; Zamora, D.B.; McElwee, M.; Vijayakrishnan, S.; Richardson, C.A.; et al. Host Vesicle Fusion Protein VAPB Contributes to the Nuclear Egress Stage of Herpes Simplex Virus Type-1 (HSV-1) Replication. Cells 2019, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Votteler, J.; Sundquist, W.I. Virus budding and the ESCRT pathway. Cell Host Microbe 2013, 14, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Vietri, M.; Radulovic, M.; Stenmark, H. The many functions of ESCRTs. Nat. Rev. Mol. Cell Biol. 2020, 21, 25–42. [Google Scholar] [CrossRef]
- Crump, C.M.; Yates, C.; Minson, T. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 2007, 81, 7380–7387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandon, R.; AuCoin, D.P.; Mocarski, E.S. Human cytomegalovirus exploits ESCRT machinery in the process of virion maturation. J. Virol. 2009, 83, 10797–10807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharkwal, H.; Smith, C.G.; Wilson, D.W. Blocking ESCRT-mediated envelopment inhibits microtubule-dependent trafficking of alphaherpesviruses in vitro. J. Virol. 2014, 88, 14467–14478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pawliczek, T.; Crump, C.M. Herpes simplex virus type 1 production requires a functional ESCRT-III complex but is independent of TSG101 and ALIX expression. J. Virol. 2009, 83, 11254–11264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vietri, M.; Schink, K.O.; Campsteijn, C.; Wegner, C.S.; Schultz, S.W.; Christ, L.; Thoresen, S.B.; Brech, A.; Raiborg, C.; Stenmark, H. Spastin and ESCRT-III coordinate mitotic spindle disassembly and nuclear envelope sealing. Nature 2015, 522, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Denais, C.M.; Gilbert, R.M.; Isermann, P.; McGregor, A.L.; te Lindert, M.; Weigelin, B.; Davidson, P.M.; Friedl, P.; Wolf, K.; Lammerding, J. Nuclear envelope rupture and repair during cancer cell migration. Science 2016, 352, 353–358. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.P.; Liu, P.T.; Kung, H.N.; Su, M.T.; Chua, H.H.; Chang, Y.H.; Chang, C.W.; Tsai, C.H.; Liu, F.T.; Chen, M.R. The ESCRT machinery is recruited by the viral BFRF1 protein to the nucleus-associated membrane for the maturation of Epstein-Barr Virus. PLoS Pathog. 2012, 8, e1002904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.P.; Liu, G.T.; Kung, H.N.; Liu, P.T.; Liao, Y.T.; Chow, L.P.; Chang, L.S.; Chang, Y.H.; Chang, C.W.; Shu, W.C.; et al. The Ubiquitin Ligase Itch and Ubiquitination Regulate BFRF1-Mediated Nuclear Envelope Modification for Epstein-Barr Virus Maturation. J. Virol. 2016, 90, 8994–9007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leis, J.; Luan, C.H.; Audia, J.E.; Dunne, S.F.; Heath, C.M. Ilaprazole and other novel prazole-based compounds that bind Tsg101 inhibit viral budding of HSV-1/2 and HIV from cells. J. Virol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.P.; Stahelin, R.V. The Ebola virus matrix protein VP40 selectively induces vesiculation from phosphatidylserine-enriched membranes. J. Biol. Chem. 2014, 289, 33590–33597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Effect of the pseudorabies virus US3 protein on nuclear membrane localization of the UL34 protein and virus egress from the nucleus. J. Gen. Virol 2001, 82, 2363–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryckman, B.J.; Roller, R.J. Herpes simplex virus type 1 primary envelopment: UL34 protein modification and the US3-UL34 catalytic relationship. J. Virol. 2004, 78, 399–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, A.; Oda, S.; Watanabe, M.; Oyama, M.; Kozuka-Hata, H.; Koyanagi, N.; Maruzuru, Y.; Arii, J.; Kawaguchi, Y. Roles of the Phosphorylation of Herpes Simplex Virus 1 UL51 at a Specific Site in Viral Replication and Pathogenicity. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riva, L.; Thiry, M.; Lebrun, M.; L’Homme, L.; Piette, J.; Sadzot-Delvaux, C. Deletion of the ORF9p acidic cluster impairs the nuclear egress of varicella-zoster virus capsids. J. Virol. 2015, 89, 2436–2441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, C.C.; Wisner, T.W.; Hannah, B.P.; Eisenberg, R.J.; Cohen, G.H.; Johnson, D.C. Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J. Virol. 2009, 83, 11847–11856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arii, J.; Kawaguchi, Y. The Role of HSV Glycoproteins in Mediating Cell Entry. Adv. Exp. Med. Biol. 2018, 1045, 3–21. [Google Scholar] [CrossRef]
- Ott, M.; Tascher, G.; Hassdenteufel, S.; Zimmermann, R.; Haas, J.; Bailer, S.M. Functional characterization of the essential tail anchor of the herpes simplex virus type 1 nuclear egress protein pUL34. J. Gen. Virol. 2011, 92, 2734–2745. [Google Scholar] [CrossRef] [PubMed]
- Schuster, F.; Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Structural determinants for nuclear envelope localization and function of pseudorabies virus pUL34. J. Virol. 2012, 86, 2079–2088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirohata, Y.; Arii, J.; Liu, Z.; Shindo, K.; Oyama, M.; Kozuka-Hata, H.; Sagara, H.; Kato, A.; Kawaguchi, Y. Herpes Simplex Virus 1 Recruits CD98 Heavy Chain and beta1 Integrin to the Nuclear Membrane for Viral De-Envelopment. J. Virol. 2015, 89, 7799–7812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Kato, A.; Oyama, M.; Kozuka-Hata, H.; Arii, J.; Kawaguchi, Y. Role of Host Cell p32 in Herpes Simplex Virus 1 De-Envelopment during Viral Nuclear Egress. J. Virol. 2015, 89, 8982–8998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohgimoto, S.; Tabata, N.; Suga, S.; Nishio, M.; Ohta, H.; Tsurudome, M.; Komada, H.; Kawano, M.; Watanabe, N.; Ito, Y. Molecular characterization of fusion regulatory protein-1 (FRP-1) that induces multinucleated giant cell formation of monocytes and HIV gp160-mediated cell fusion. FRP-1 and 4F2/CD98 are identical molecules. J. Immunol. 1995, 155, 3585–3592. [Google Scholar] [PubMed]
- Okamoto, K.; Tsurudome, M.; Ohgimoto, S.; Kawano, M.; Nishio, M.; Komada, H.; Ito, M.; Sakakura, Y.; Ito, Y. An anti-fusion regulatory protein-1 monoclonal antibody suppresses human parainfluenza virus type 2-induced cell fusion. J. Gen. Virol. 1997, 78, 83–89. [Google Scholar] [CrossRef] [Green Version]
- Ohta, H.; Tsurudome, M.; Matsumura, H.; Koga, Y.; Morikawa, S.; Kawano, M.; Kusugawa, S.; Komada, H.; Nishio, M.; Ito, Y. Molecular and biological characterization of fusion regulatory proteins (FRPs): Anti-FRP mAbs induced HIV-mediated cell fusion via an integrin system. EMBO J. 1994, 13, 2044–2055. [Google Scholar] [CrossRef]
- Ito, Y.; Komada, H.; Kusagawa, S.; Tsurudome, M.; Matsumura, H.; Kawano, M.; Ohta, H.; Nishio, M. Fusion regulation proteins on the cell surface: Isolation and characterization of monoclonal antibodies which enhance giant polykaryocyte formation in Newcastle disease virus-infected cell lines of human origin. J. Virol. 1992, 66, 5999–6007. [Google Scholar] [CrossRef] [Green Version]
- Maeda, F.; Arii, J.; Hirohata, Y.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. Herpes Simplex Virus 1 UL34 Protein Regulates the Global Architecture of the Endoplasmic Reticulum in Infected Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Hassinger, L.; Thomson, T.; Ding, B.; Ashley, J.; Hassinger, W.; Budnik, V. Lamin Mutations Accelerate Aging via Defective Export of Mitochondrial mRNAs through Nuclear Envelope Budding. Curr. Biol. 2016, 26, 2052–2059. [Google Scholar] [CrossRef] [Green Version]
- Maric, M.; Shao, J.; Ryan, R.J.; Wong, C.S.; Gonzalez-Alegre, P.; Roller, R.J. A functional role for TorsinA in herpes simplex virus 1 nuclear egress. J. Virol. 2011, 85, 9667–9679. [Google Scholar] [CrossRef] [Green Version]
- Jokhi, V.; Ashley, J.; Nunnari, J.; Noma, A.; Ito, N.; Wakabayashi-Ito, N.; Moore, M.J.; Budnik, V. Torsin mediates primary envelopment of large ribonucleoprotein granules at the nuclear envelope. Cell Rep. 2013, 3, 988–995. [Google Scholar] [CrossRef] [Green Version]
- Rampello, A.J.; Prophet, S.M.; Schlieker, C. The Role of Torsin AAA+ Proteins in Preserving Nuclear Envelope Integrity and Safeguarding Against Disease. Biomolecules 2020, 10, 468. [Google Scholar] [CrossRef] [Green Version]
- Laudermilch, E.; Schlieker, C. Torsin ATPases: Structural insights and functional perspectives. Curr. Opin. Cell Biol. 2016, 40, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Brown, R.S.; Zhao, C.; Chase, A.R.; Wang, J.; Schlieker, C. The mechanism of Torsin ATPase activation. Proc. Natl. Acad. Sci. USA 2014, 111, E4822–E4831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, B.A.; Demircioglu, F.E.; Chen, J.Z.; Ingram, J.; Ploegh, H.L.; Schwartz, T.U. How lamina-associated polypeptide 1 (LAP1) activates Torsin. Elife 2014, 3, e03239. [Google Scholar] [CrossRef] [PubMed]
- Gyorgy, B.; Cruz, L.; Yellen, D.; Aufiero, M.; Alland, I.; Zhang, X.; Ericsson, M.; Fraefel, C.; Li, Y.C.; Takeda, S.; et al. Mutant torsinA in the heterozygous DYT1 state compromises HSV propagation in infected neurons and fibroblasts. Sci. Rep. 2018, 8, 2324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, E.M.; Brown, R.S.; Laudermilch, E.; Tsai, P.L.; Schlieker, C. The Torsin Activator LULL1 Is Required for Efficient Growth of Herpes Simplex Virus 1. J. Virol. 2015, 89, 8444–8452. [Google Scholar] [CrossRef] [Green Version]
- Goodchild, R.E.; Kim, C.E.; Dauer, W.T. Loss of the dystonia-associated protein torsinA selectively disrupts the neuronal nuclear envelope. Neuron 2005, 48, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Liang, C.C.; Tanabe, L.M.; Jou, S.; Chi, F.; Dauer, W.T. TorsinA hypofunction causes abnormal twisting movements and sensorimotor circuit neurodegeneration. J. Clin. Investig. 2014, 124, 3080–3092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grillet, M.; Dominguez Gonzalez, B.; Sicart, A.; Pottler, M.; Cascalho, A.; Billion, K.; Hernandez Diaz, S.; Swerts, J.; Naismith, T.V.; Gounko, N.V.; et al. Torsins Are Essential Regulators of Cellular Lipid Metabolism. Dev. Cell 2016, 38, 235–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arii, J.; Goto, H.; Suenaga, T.; Oyama, M.; Kozuka-Hata, H.; Imai, T.; Minowa, A.; Akashi, H.; Arase, H.; Kawaoka, Y.; et al. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 2010, 467, 859–862. [Google Scholar] [CrossRef] [PubMed]
- Arii, J.; Hirohata, Y.; Kato, A.; Kawaguchi, Y. Nonmuscle myosin heavy chain IIb mediates herpes simplex virus 1 entry. J. Virol. 2015, 89, 1879–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A.; Uehori, J.; Arase, N.; Shiratori, I.; Tanaka, S.; Kawaguchi, Y.; et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laudermilch, E.; Tsai, P.L.; Graham, M.; Turner, E.; Zhao, C.; Schlieker, C. Dissecting Torsin/cofactor function at the nuclear envelope: A genetic study. Mol. Biol. Cell 2016, 27, 3964–3971. [Google Scholar] [CrossRef] [PubMed]
- Webster, B.M.; Colombi, P.; Jager, J.; Lusk, C.P. Surveillance of nuclear pore complex assembly by ESCRT-III/Vps4. Cell 2014, 159, 388–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webster, B.M.; Thaller, D.J.; Jager, J.; Ochmann, S.E.; Borah, S.; Lusk, C.P. Chm7 and Heh1 collaborate to link nuclear pore complex quality control with nuclear envelope sealing. EMBO J. 2016, 35, 2447–2467. [Google Scholar] [CrossRef]
- Olmos, Y.; Hodgson, L.; Mantell, J.; Verkade, P.; Carlton, J.G. ESCRT-III controls nuclear envelope reformation. Nature 2015, 522, 236–239. [Google Scholar] [CrossRef] [Green Version]
- Raab, M.; Gentili, M.; de Belly, H.; Thiam, H.R.; Vargas, P.; Jimenez, A.J.; Lautenschlaeger, F.; Voituriez, R.; Lennon-Dumenil, A.M.; Manel, N.; et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science 2016, 352, 359–362. [Google Scholar] [CrossRef]
- Eriksson, M.; Brown, W.T.; Gordon, L.B.; Glynn, M.W.; Singer, J.; Scott, L.; Erdos, M.R.; Robbins, C.M.; Moses, T.Y.; Berglund, P.; et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003, 423, 293–298. [Google Scholar] [CrossRef] [Green Version]
- De Sandre-Giovannoli, A.; Bernard, R.; Cau, P.; Navarro, C.; Amiel, J.; Boccaccio, I.; Lyonnet, S.; Stewart, C.L.; Munnich, A.; Le Merrer, M.; et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003, 300, 2055. [Google Scholar] [CrossRef] [PubMed]
- Arii, J.; Maeda, F.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Mori, Y.; Kawaguchi, Y. ESCRT-III controls nuclear envelope deformation induced by progerin. Sci. Rep. 2020, 10, 18877. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Conservation | Function |
|---|---|---|
| NEC (UL31/UL34) | Herpesviridae | Vesicle formation via hexagonal lattice [63]. Incorporation of capsid [13,14]. Dissociation of lamins [29]. Recruitment of ESCRT-III [80]. De-envelopment [81]. |
| UL13 | Herpesviridae | Dissociation of lamins [82]. (Weak in Alphaherpesvirinae). |
| Us3 | Alphaherpesvirinae | Dissociation of lamins [33]. Promote de-envelopment [83]. Phosphorylate UL34 [84]. Phosphorylate gB [85,86]. Phosphorylate UL31 [87]. |
| ICP34.5 | HSV-1 and HSV-2 | Dissociation of lamins via PKC [42]. |
| CVSC (UL17/UL25) | Herpesviridae | Link between NEC and capsid [55,56]. |
| UL47 | Alphaherpesvirinae | Promote primary envelopment [78]. (Not observed in PRV [88,89]). |
| ICP22 | Alphaherpesvirinae | Promote primary envelopment [79]. |
| gB and gH/gL | Herpesviridae | Redundantly promote de-envelopment [90]. (Not observed in PRV [91]). |
| UL51 | Herpesviridae | Promote de-envelopment [92]. (Not observed in PRV [93]). |
| Protein | Interactor | Function | Drosophila RNP |
|---|---|---|---|
| Lamins | NEC, Us3, UL13 | Prevent nuclear egress [29]. | Yes [6,122] |
| PKC family | NEC | Dissociate lamins [32]. | Yes [6] |
| p32 | ICP34.5, UL47 | Recruit PKC [42]. Promote de-envelopment [116]. | - |
| PKD | - | Promote nuclear egress indirectly [67]. | - |
| VAPB | - | Promote nuclear egress [94]. | - |
| ESCRT-III | NEC | Mediate scission of INM [80]. | Yes [80] |
| ALIX | NEC | Recruit ESCRT-III to INM [80]. | - |
| TSG101 | - | Nuclear egress [105]. | |
| CD98hc β1 integrin | NEC, gB, gH | Promote de-envelopment [115]. (Modulate fusion activity?). | - |
| Torsins | - | Promote de-envelopment indirectly [123]. | Yes [124] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arii, J. Host and Viral Factors Involved in Nuclear Egress of Herpes Simplex Virus 1. Viruses 2021, 13, 754. https://doi.org/10.3390/v13050754
Arii J. Host and Viral Factors Involved in Nuclear Egress of Herpes Simplex Virus 1. Viruses. 2021; 13(5):754. https://doi.org/10.3390/v13050754
Chicago/Turabian StyleArii, Jun. 2021. "Host and Viral Factors Involved in Nuclear Egress of Herpes Simplex Virus 1" Viruses 13, no. 5: 754. https://doi.org/10.3390/v13050754
APA StyleArii, J. (2021). Host and Viral Factors Involved in Nuclear Egress of Herpes Simplex Virus 1. Viruses, 13(5), 754. https://doi.org/10.3390/v13050754