
Structural Insight into Non-Enveloped Virus Binding to Glycosaminoglycan Receptors: A Review


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
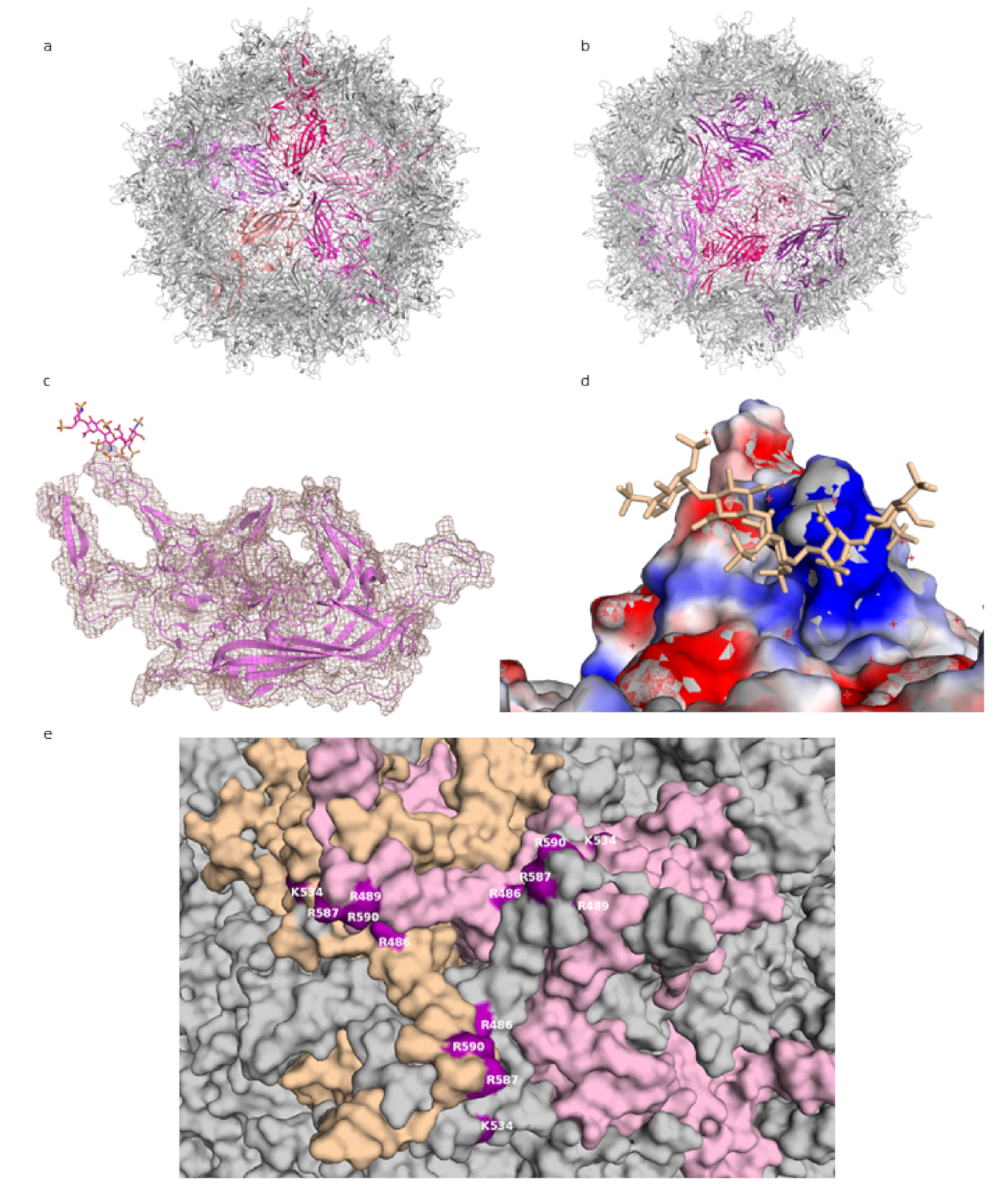
2. Adeno-Associated Virus

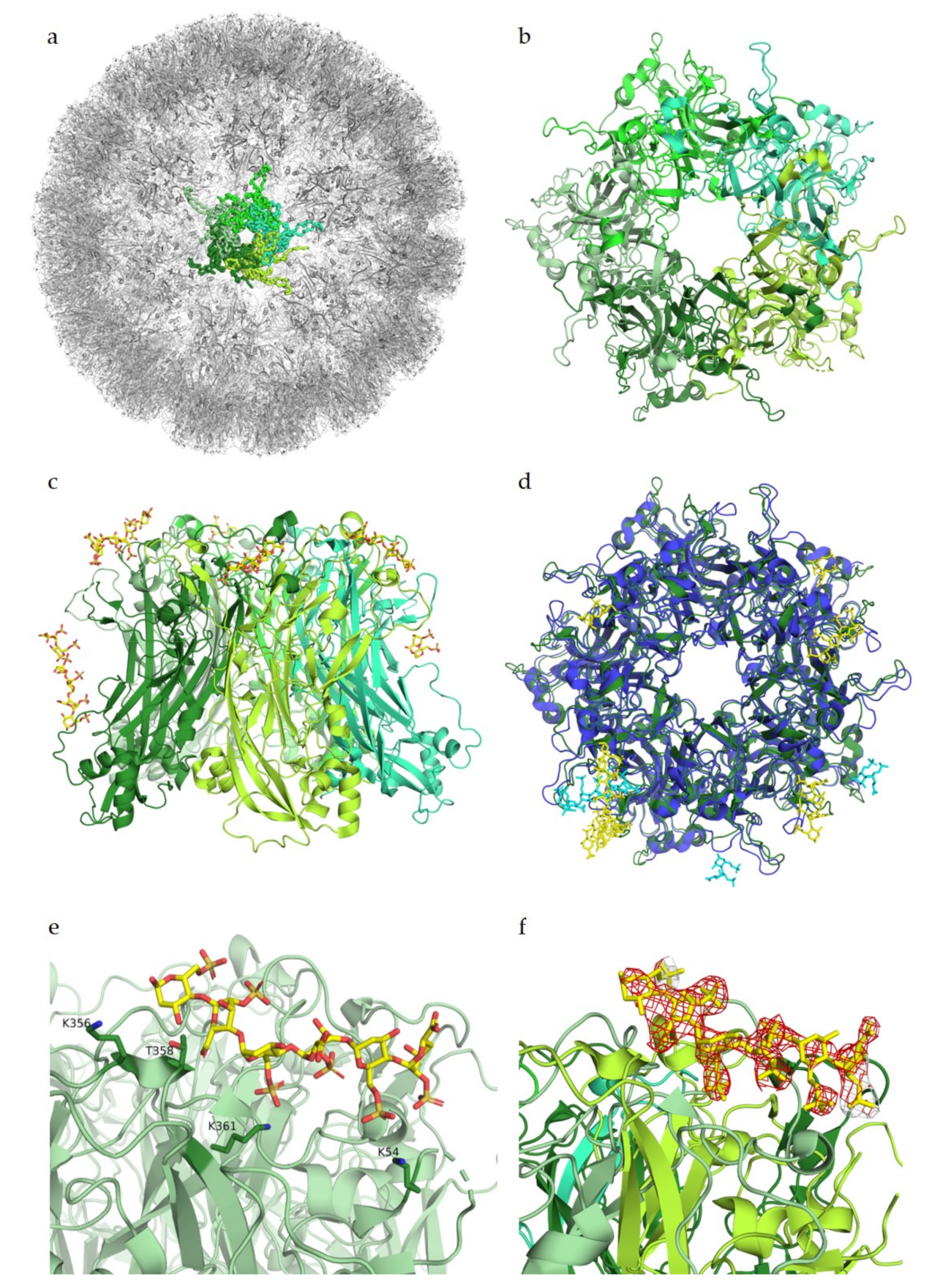
3. Human Papillomavirus
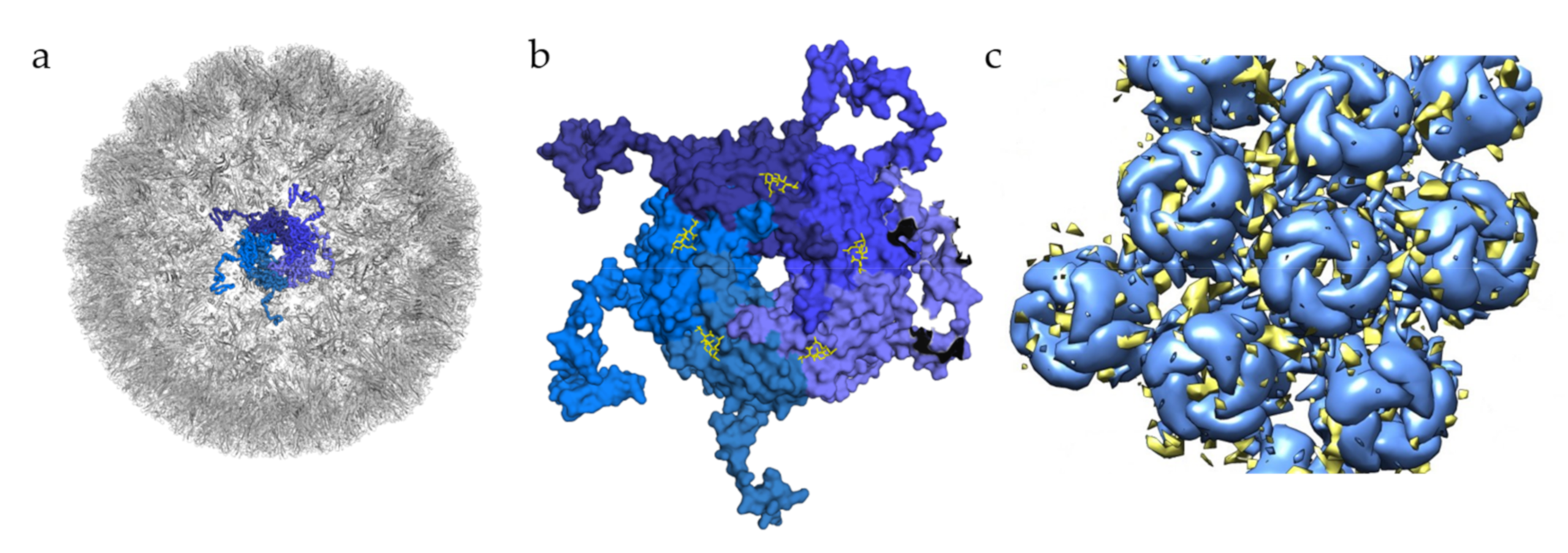
4. Polyomavirus

5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suenaga, T.; Arase, H. Viral Interactions with Glycans. Glycosci. Biol. Med. 2014, 785, 785–794. [Google Scholar]
- Gandhi, N.S.; Mancera, R.L. The Structure of Glycosaminoglycans and their Interactions with Proteins. Chem. Biol. Drug Des. 2008, 72, 455–482. [Google Scholar] [CrossRef] [PubMed]
- Rabenstein, D.L. Heparin and heparan sulfate: Structure and function. Nat. Prod. Rep. 2002, 19, 312–331. [Google Scholar] [CrossRef]
- Gallagher, J.T.; Walker, A. Molecular distinctions between heparan sulphate and heparin. Analysis of sulphation patterns indicates that heparan sulphate and heparin are separate families of N-sulphated polysaccharides. Biochem. J. 1985, 230, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Mikami, T.; Kitagawa, H. Biosynthesis and function of chondroitin sulfate. Biochim. Biophys. Acta (BBA) Gen. Subj. 2013, 1830, 4719–4733. [Google Scholar] [CrossRef]
- Trowbridge, J.M.; Gallo, R.L. Dermatan sulfate: New functions from an old glycosaminoglycan. Glycobiology 2002, 12, 117R–125R. [Google Scholar] [CrossRef]
- Dechecchi, M.C.; Melotti, P.; Bonizzato, A.; Santacatterina, M.; Chilosi, M.; Cabrini, G. Heparan Sulfate Glycosaminoglycans Are Receptors Sufficient To Mediate the Initial Binding of Adenovirus Types 2 and 5. J. Virol. 2001, 75, 8772–8780. [Google Scholar] [CrossRef] [Green Version]
- Khan, A.G.; Pichler, J.; Rosemann, A.; Blaas, D. Human Rhinovirus Type 54 Infection via Heparan Sulfate Is Less Efficient and Strictly Dependent on Low Endosomal pH. J. Virol. 2007, 81, 4625–4632. [Google Scholar] [CrossRef] [Green Version]
- Cagno, V.; Tseligka, E.D.; Jones, S.T.; Tapparel, C. Heparan Sulfate Proteoglycans and Viral Attachment: True Receptors or Adaptation Bias? Viruses 2019, 11, 596. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Bu, W.; Bhatia, S.; Hare, J.; Somasundaram, T.; Azzi, A.; Chapman, M.S. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. USA 2002, 99, 10405–10410. [Google Scholar] [CrossRef] [Green Version]
- Kern, A.; Schmidt, K.; Leder, C.; Müller, O.J.; Wobus, C.E.; Bettinger, K.; Von Der Lieth, C.W.; King, J.A.; Kleinschmidt, J.A. Identification of a Heparin-Binding Motif on Adeno-Associated Virus Type 2 Capsids. J. Virol. 2003, 77, 11072–11081. [Google Scholar] [CrossRef] [Green Version]
- Ng, R.; Govindasamy, L.; Gurda, B.L.; McKenna, R.; Kozyreva, O.G.; Samulski, R.J.; Parent, K.N.; Baker, T.S.; Agbandje-McKenna, M. Structural Characterization of the Dual Glycan Binding Adeno-Associated Virus Serotype 6. J. Virol. 2010, 84, 12945–12957. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Lerch, T.F.; Meyer, N.L.; Chapman, M.S. Structure–function analysis of receptor-binding in adeno-associated virus serotype 6 (AAV-6). Virology 2011, 420, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Pillay, S.; Meyer, N.L.; Puschnik, A.S.; Davulcu, O.; Diep, J.; Ishikawa, Y.; Jae, L.T.; Wosen, J.E.; Nagamine, C.M.; Chapman, M.S.; et al. An essential receptor for adeno-associated virus infection. Nat. Cell Biol. 2016, 530, 108–112. [Google Scholar] [CrossRef]
- Xie, Q.; Spilman, M.; Meyer, N.L.; Lerch, T.F.; Stagg, S.M.; Chapman, M.S. Electron microscopy analysis of a disaccharide analog complex reveals receptor interactions of adeno-associated virus. J. Struct. Biol. 2013, 184, 129–135. [Google Scholar] [CrossRef] [Green Version]
- Hurtley, S.M. Continuing the resolution revolution. Science 2018, 360, 280–282. [Google Scholar] [CrossRef]
- Levy, H.C.; Bowman, V.D.; Govindasamy, L.; McKenna, R.; Nash, K.; Warrington, K.; Chen, W.; Muzyczka, N.; Yan, X.; Baker, T.S.; et al. Heparin binding induces conformational changes in Adeno-associated virus serotype 2. J. Struct. Biol. 2009, 165, 146–156. [Google Scholar] [CrossRef] [Green Version]
- O’Donnell, J.; Taylor, K.A.; Chapman, M.S. Adeno-associated virus-2 and its primary cellular receptor—Cryo-EM structure of a heparin complex. Virology 2009, 385, 434–443. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Spear, J.M.; Noble, A.J.; Sousa, D.R.; Meyer, N.L.; Davulcu, O.; Zhang, F.; Linhardt, R.J.; Stagg, S.M.; Chapman, M.S. The 2.8 Å Electron Microscopy Structure of Adeno-Associated Virus-DJ Bound by a Heparinoid Pentasaccharide. Mol. Ther. Methods Clin. Dev. 2017, 5, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Xie, Q.; Yoshioka, C.K.; Chapman, M.S. Adeno-Associated Virus (AAV-DJ)—Cryo-EM Structure at 1.56 Å Resolution. Viruses 2020, 12, 1194. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Tai, P.W.L.; Gao, G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019, 18, 358–378. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L.L.C. The {PyMOL} Molecular Graphics System, Version~1.8. 2015. Available online: https://pymol.org/2/ (accessed on 14 April 2021).
- Bzhalava, D.; Guan, P.; Franceschi, S.; Dillner, J.; Clifford, G. A systematic review of the prevalence of mucosal and cutaneous human papillomavirus types. Virology 2013, 445, 224–231. [Google Scholar] [CrossRef] [Green Version]
- Lei, J.; Ploner, A.; Elfström, K.M.; Wang, J.; Roth, A.; Fang, F.; Sundström, K.; Dillner, J.; Sparén, P. HPV Vaccination and the Risk of Invasive Cervical Cancer. N. Engl. J. Med. 2020, 383, 1340–1348. [Google Scholar] [CrossRef]
- Crosbie, E.J.; Einstein, M.H.; Franceschi, S.; Kitchener, H.C. Human papillomavirus and cervical cancer. Lancet 2013, 382, 889–899. [Google Scholar] [CrossRef]
- Shafti-Keramat, S.; Handisurya, A.; Kriehuber, E.; Meneguzzi, G.; Slupetzky, K.; Kirnbauer, R. Different Heparan Sulfate Proteoglycans Serve asCellular Receptors for HumanPapillomaviruses. J. Virol. 2003, 77, 13125–13135. [Google Scholar] [CrossRef] [Green Version]
- Buck, C.B.; Thompson, C.D.; Roberts, J.N.; Müller, M.; Lowy, D.R.; Schiller, J.T. Carrageenan Is a Potent Inhibitor of Papillomavirus Infection. PLOS Pathog. 2006, 2, e69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rommel, O.; Dillner, J.; Fligge, C.; Bergsdorf, C.; Wang, X.; Selinka, H.-C.; Sapp, M. Heparan sulfate proteoglycans interact exclusively with conformationally intact HPV L1 assemblies: Basis for a virus-like particle ELISA. J. Med. Virol. 2005, 75, 114–121. [Google Scholar] [CrossRef]
- Bousarghin, L.; Touzé, A.; Combita-Rojas, A.-L.; Coursaget, P. Positively charged sequences of human papillomavirus type 16 capsid proteins are sufficient to mediate gene transfer into target cells via the heparan sulfate receptor. J. Gen. Virol. 2003, 84, 157–164. [Google Scholar] [CrossRef]
- Sun, J.; Yu, J.S.; Jin, S.; Zha, X.; Wu, Y.; Yu, Z. Interaction of synthetic HPV-16 capsid peptides with heparin: Thermodynamic parameters and binding mechanism. J. Phys. Chem. B 2010, 114, 9854–9861. [Google Scholar] [CrossRef]
- Dasgupta, J.; Bienkowska-Haba, M.; Ortega, M.E.; Patel, H.D.; Bodevin, S.; Spillmann, D.; Bishop, B.; Sapp, M.; Chen, X.S. Structural Basis of Oligosaccharide Receptor Recognition by Human Papillomavirus. J. Biol. Chem. 2011, 286, 2617–2624. [Google Scholar] [CrossRef] [Green Version]
- Guan, J.; Bywaters, S.M.; Brendle, S.A.; Ashley, R.E.; Makhov, A.M.; Conway, J.F.; Christensen, N.D.; Hafenstein, S. Cryoelectron Microscopy Maps of Human Papillomavirus 16 Reveal L2 Densities and Heparin Binding Site. Structure 2017, 25, 253–263. [Google Scholar] [CrossRef] [Green Version]
- Maginnis, M.S. Human Polyomaviruses (Papillomaviridae). In Encyclopedia of Virology, 4th ed.; Bamford, D.H., Zuckerman, M., Eds.; Academic Press: Oxford, UK, 2021; pp. 518–527. [Google Scholar]
- Hirsch, H.H. Polyomavirus BK nephropathy: A (re-)emerging complication in renal transplantation. Am. J. Transplant. 2002, 2, 25–30. [Google Scholar] [CrossRef]
- Padgett, B.L.; Walker, D.L.; ZuRhein, G.M.; Eckroade, R.J.; Dessel, B.H. Cultivation of papova-like virus from human brain with progressive multifocal leucoencephalopathy. Lancet 1971, 1, 1257–1260. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [Green Version]
- Belnap, D.M.; Olson, N.H.; Cladel, N.M.; Newcomb, W.W.; Brown, J.C.; Kreider, J.W.; Christensen, N.D.; Baker, T.S. Conserved Features in Papillomavirus and Polyomavirus Capsids. J. Mol. Biol. 1996, 259, 249–263. [Google Scholar] [CrossRef] [Green Version]
- Stehle, T.; Yan, Y.; Benjamin, T.L.; Harrison, S.C. Structure of murine polyomavirus complexed with an oligosaccharide receptor fragment. Nat. Cell Biol. 1994, 369, 160–163. [Google Scholar] [CrossRef]
- Neu, U.; Hengel, H.; Blaum, B.S.; Schowalter, R.M.; Macejak, D.; Gilbert, M.; Wakarchuk, W.W.; Imamura, A.; Ando, H.; Kiso, M.; et al. Structures of Merkel Cell Polyomavirus VP1 Complexes Define a Sialic Acid Binding Site Required for Infection. PLOS Pathog. 2012, 8, e1002738. [Google Scholar] [CrossRef]
- Neu, U.; Allen, S.-A.A.; Blaum, B.S.; Liu, Y.; Frank, M.; Palma, A.S.; Ströh, L.J.; Feizi, T.; Peters, T.; Atwood, W.J.; et al. A Structure-Guided Mutation in the Major Capsid Protein Retargets BK Polyomavirus. PLoS Pathog. 2013, 9, e1003688. [Google Scholar] [CrossRef] [Green Version]
- Khan, Z.M.; Liu, Y.; Neu, U.; Gilbert, M.; Ehlers, B.; Feizi, T.; Stehle, T. Crystallographic and glycan microarray analysis of human polyomavirus 9 VP1 identifies N-glycolyl neuraminic acid as a receptor candidate. J. Virol. 2014, 88, 6100–6111. [Google Scholar] [CrossRef] [Green Version]
- Ströh, L.J.; Gee, G.V.; Blaum, B.S.; Dugan, A.S.; Feltkamp, M.C.W.; Atwood, W.J.; Stehle, T. Trichodysplasia spinulosa-Associated Polyomavirus Uses a Displaced Binding Site on VP1 to Engage Sialylated Glycolipids. PLoS Pathog. 2015, 11, e1005112. [Google Scholar] [CrossRef] [PubMed]
- Ströh, L.J.; Rustmeier, N.H.; Blaum, B.S.; Botsch, J.; Rößler, P.; Wedekink, F.; Lipkin, W.I.; Mishra, N.; Stehle, T. Structural Basis and Evolution of Glycan Receptor Specificities within the Polyomavirus Family. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Neu, U.; Maginnis, M.S.; Palma, A.S.; Ströh, L.J.; Nelson, C.D.; Feizi, T.; Atwood, W.J.; Stehle, T. Structure-Function Analysis of the Human JC Polyomavirus Establishes the LSTc Pentasaccharide as a Functional Receptor Motif. Cell Host Microbe 2010, 8, 309–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schowalter, R.M.; Pastrana, D.V.; Buck, C.B. Glycosaminoglycans and sialylated glycans sequentially facilitate Merkel cell polyomavirus infectious entry. PLoS Pathog. 2011, 7, e1002161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geoghegan, E.M.; Pastrana, D.V.; Schowalter, R.M.; Ray, U.; Gao, W.; Ho, M.; Pauly, G.T.; Sigano, D.M.; Kaynor, C.; Cahir-McFarland, E.; et al. Infectious Entry and Neutralization of Pathogenic JC Polyomaviruses. Cell Rep. 2017, 21, 1169–1179. [Google Scholar] [CrossRef] [Green Version]
- Liddington, R.C.; Yan, Y.; Moulai, J.; Sahli, R.; Benjamin, T.L.; Harrison, S.C. Structure of simian virus 40 at 3.8-A resolution. Nature 1991, 354, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Hurdiss, D.L.; Frank, M.; Snowden, J.S.; Macdonald, A.; Ranson, N.A. The Structure of an Infectious Human Polyomavirus and Its Interactions with Cellular Receptors. Structure 2018, 26, 839–847. [Google Scholar] [CrossRef] [Green Version]
- Bayer, N.J.; Januliene, D.; Zocher, G.; Stehle, T.; Moeller, A.; Blaum, B.S. Structure of Merkel Cell Polyomavirus Capsid and Interaction with Its Glycosaminoglycan Attachment Receptor. J. Virol. 2020, 94, 1–17. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Ilca, S.L.; Kotecha, A.; Sun, X.; Poranen, M.M.; Stuart, D.I.; Huiskonen, J.T. Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat. Commun. 2015, 6, 4–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sorin, M.N.; Kuhn, J.; Stasiak, A.C.; Stehle, T. Structural Insight into Non-Enveloped Virus Binding to Glycosaminoglycan Receptors: A Review. Viruses 2021, 13, 800. https://doi.org/10.3390/v13050800
Sorin MN, Kuhn J, Stasiak AC, Stehle T. Structural Insight into Non-Enveloped Virus Binding to Glycosaminoglycan Receptors: A Review. Viruses. 2021; 13(5):800. https://doi.org/10.3390/v13050800
Chicago/Turabian StyleSorin, Marie N., Jasmin Kuhn, Aleksandra C. Stasiak, and Thilo Stehle. 2021. "Structural Insight into Non-Enveloped Virus Binding to Glycosaminoglycan Receptors: A Review" Viruses 13, no. 5: 800. https://doi.org/10.3390/v13050800
APA StyleSorin, M. N., Kuhn, J., Stasiak, A. C., & Stehle, T. (2021). Structural Insight into Non-Enveloped Virus Binding to Glycosaminoglycan Receptors: A Review. Viruses, 13(5), 800. https://doi.org/10.3390/v13050800