
Mutation in a SARS-CoV-2 Haplotype from Sub-Antarctic Chile Reveals New Insights into the Spike’s Dynamics

 , , , , ,
, , , , ,  , , and
, , and 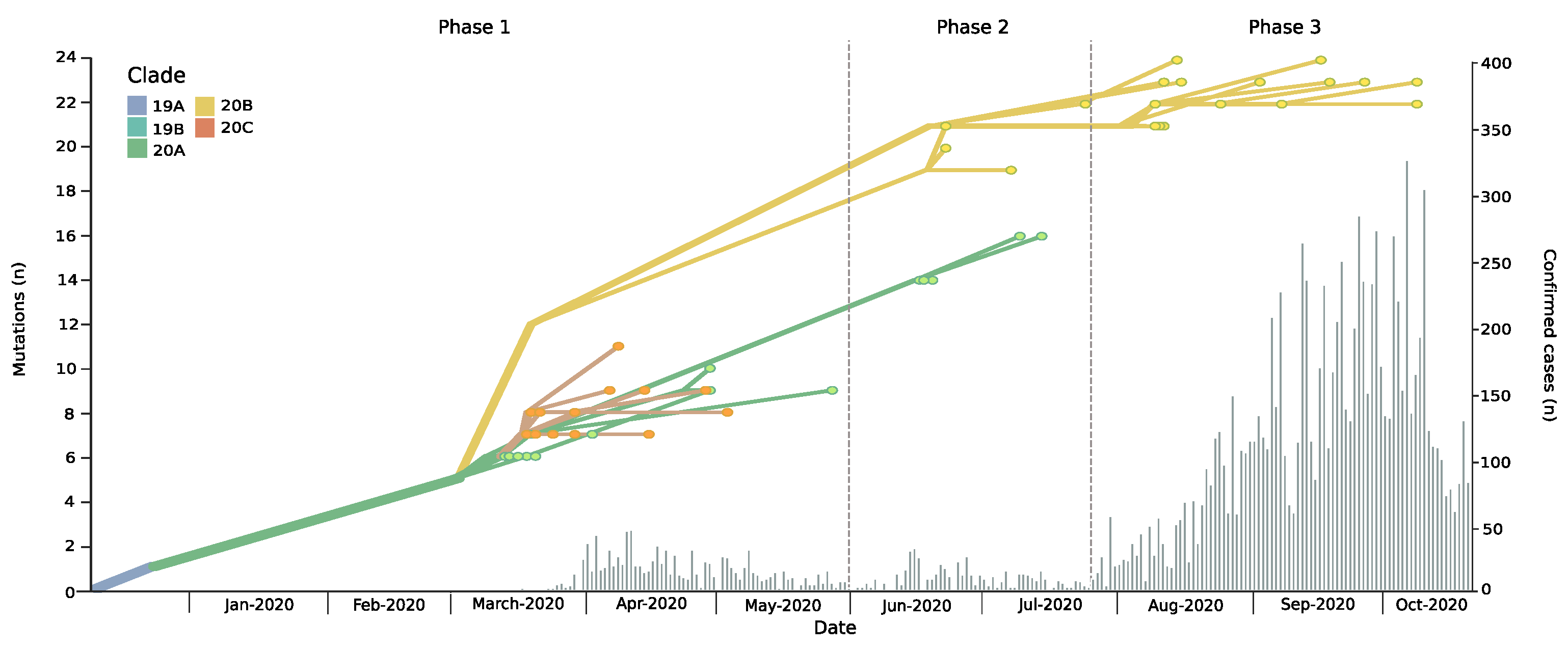{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Sites and Setting
2.2. Samples, Extraction of Genetic Material and Virus Detection
2.3. Sequencing of SARS-CoV-2 Isolates from Clinical Samples
2.4. Genome Assembly and Phylogenetic Analysis
2.5. Identification of Variants and Sequence Analysis
2.6. Protein Modeling and Molecular Dynamics Analysis
2.7. Statistical Analyses
3. Results
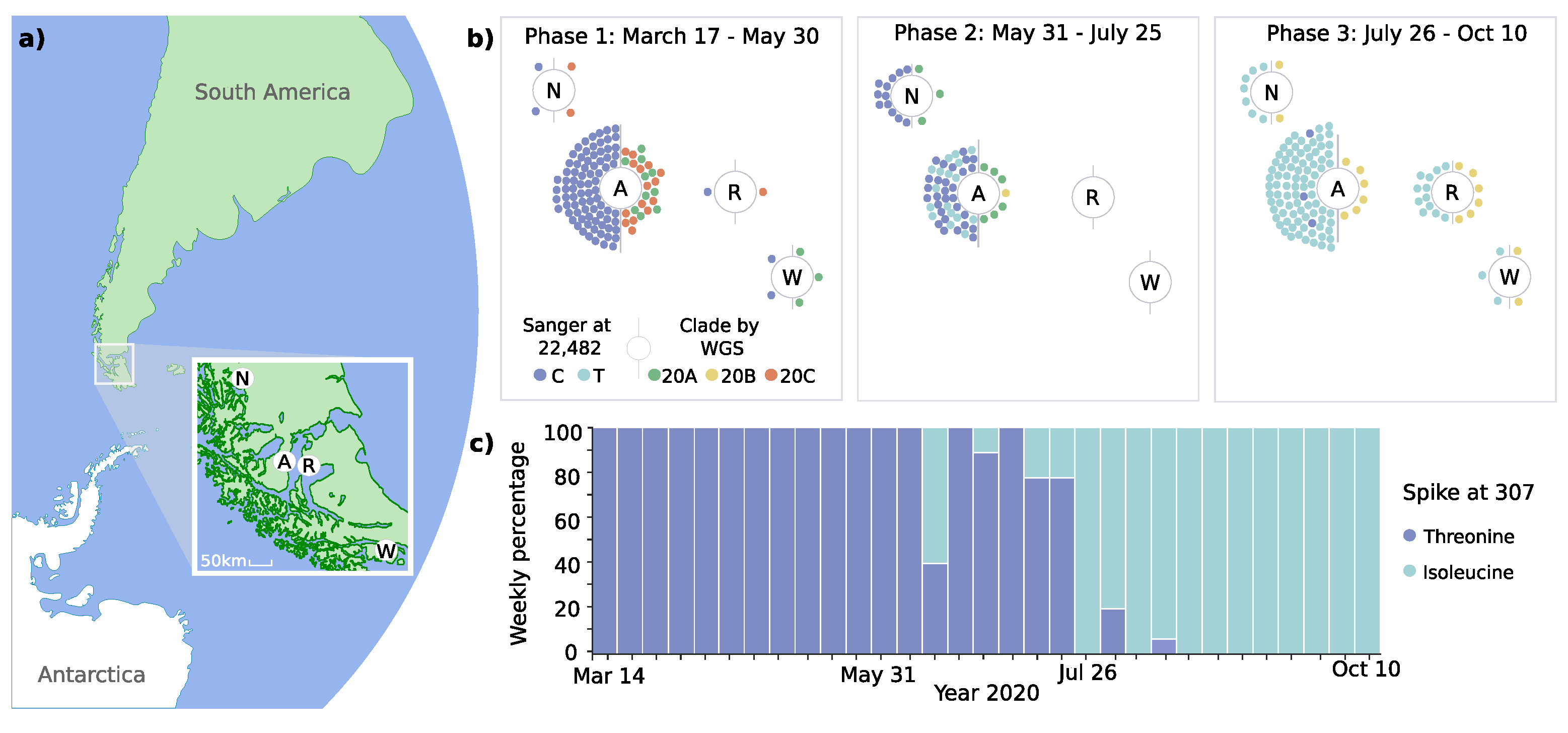
3.1. Introduction and Spread of SARS-CoV-2 in the Magallanes Region
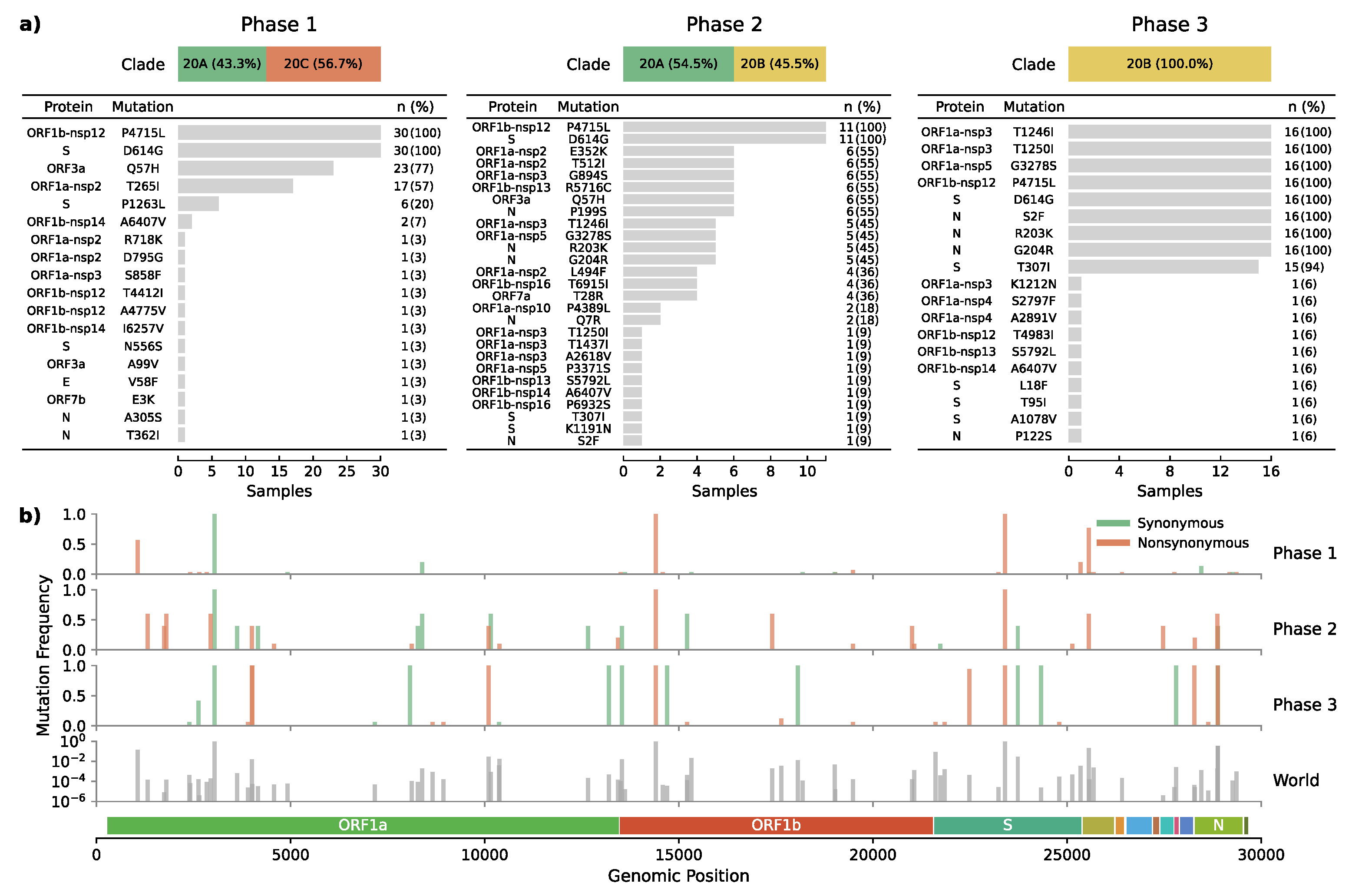
3.2. Dynamic Tracking of Variants Through Infection Spreading Events
3.3. Emergence of a Novel Haplotype with Nucleocapsid and Spike Protein Mutations
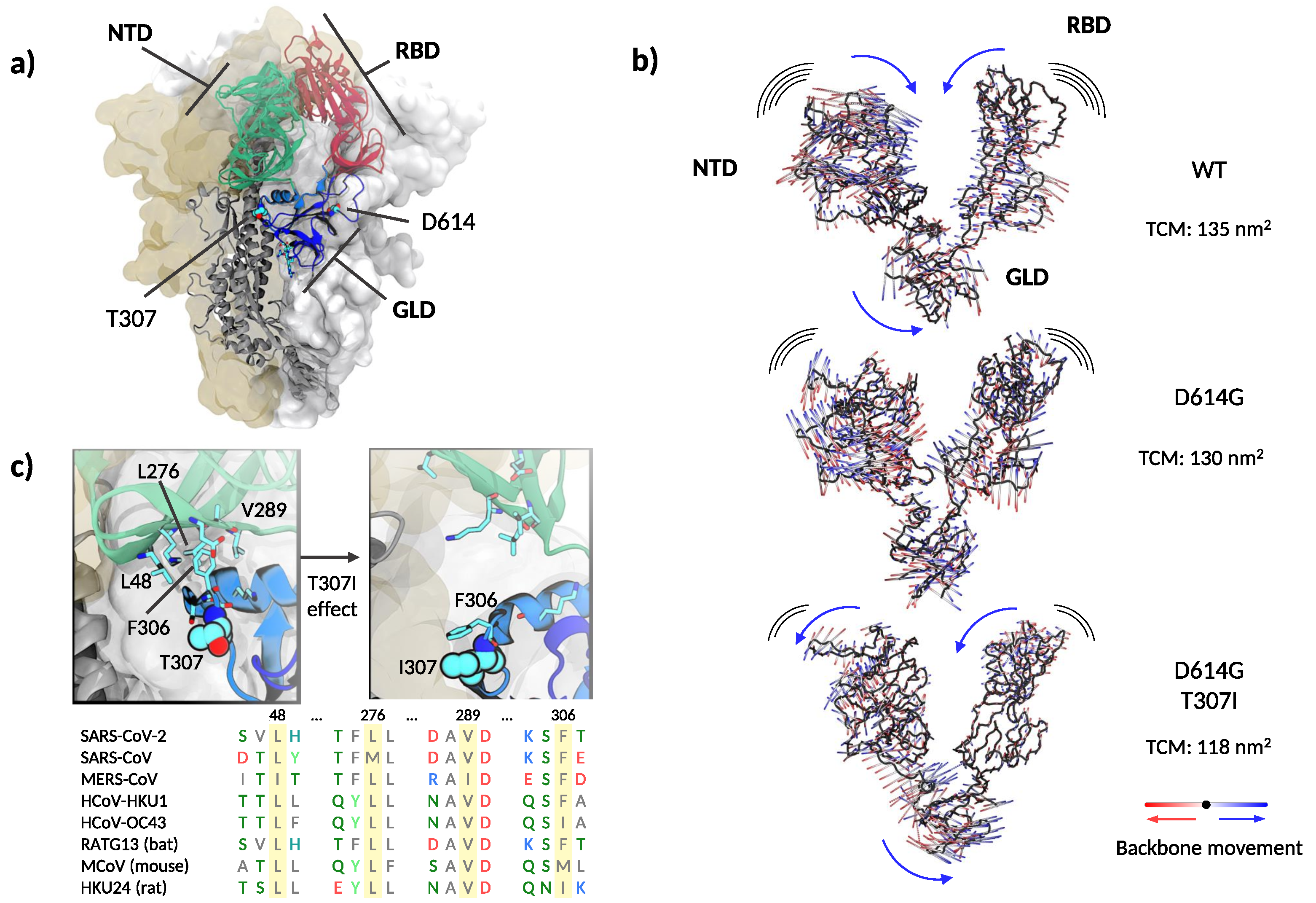
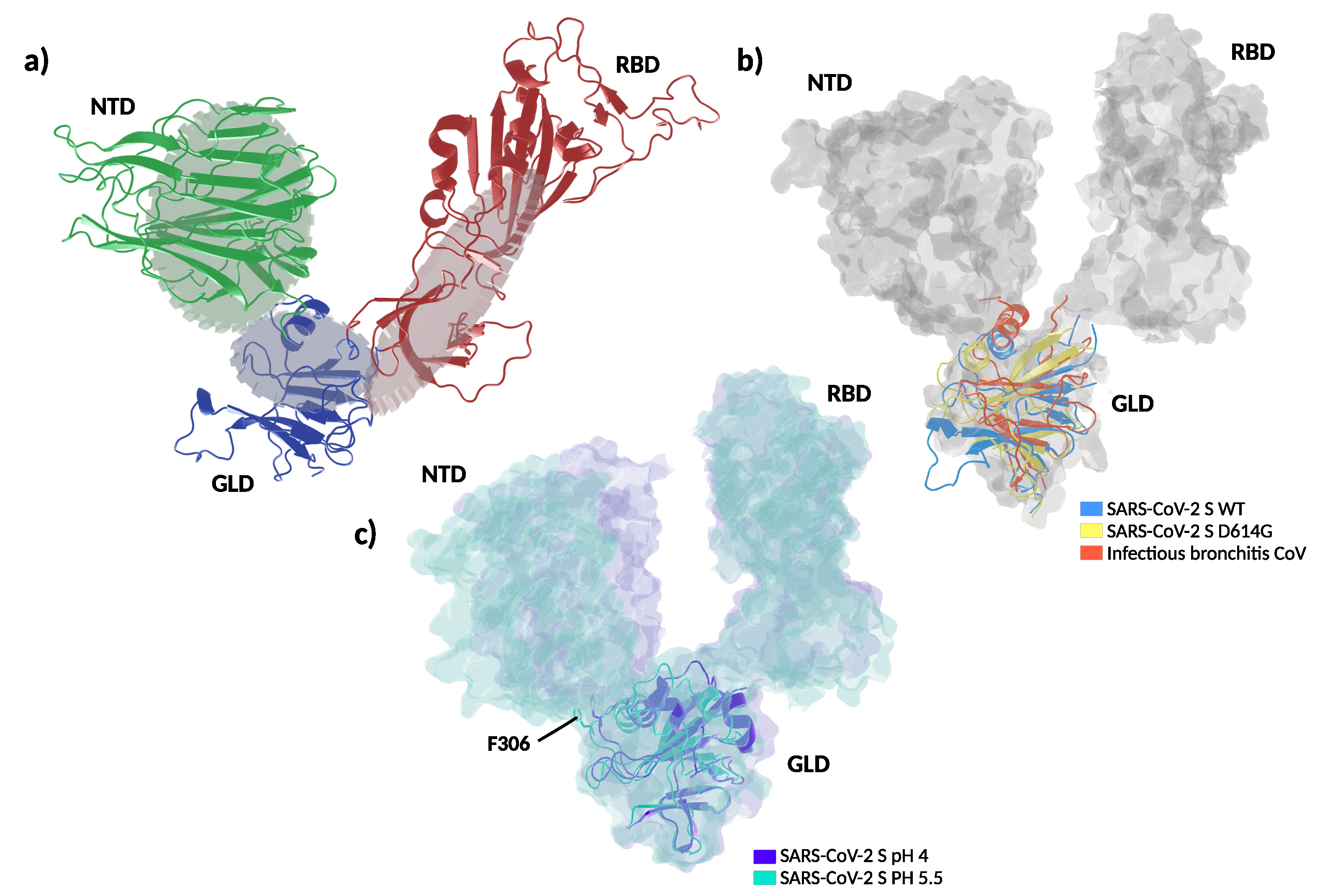
3.4. Structural and Molecular Dynamic Analysis of T307I
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| RBD | Receptor binding domain |
| NTD | N-terminal domain |
| GLD | Gear-like domain |
References
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- John Hopkins University. COVID-19 Map. Available online: https://coronavirus.jhu.edu/map.html (accessed on 25 March 2021).
- Hodcroft, E.B.; Zuber, M.; Nadeau, S.; Comas, I.; Candelas, F.G.; Consortium, S.S.; Stadler, T.; Neher, R.A. Emergence and spread of a SARS-CoV-2 variant through Europe in the summer of 2020. medRxiv 2020. [Google Scholar] [CrossRef]
- Young, B.E.; Fong, S.W.; Chan, Y.H.; Mak, T.M.; Ang, L.W.; Anderson, D.E.; Lee, C.Y.P.; Amrun, S.N.; Lee, B.; Goh, Y.S.; et al. Effects of a major deletion in the SARS-CoV-2 genome on the severity of infection and the inflammatory response: An observational cohort study. Lancet 2020, 396, 603–611. [Google Scholar] [CrossRef]
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2020, 592, 116–121. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Loman, N.; Pybus, O.; amd Jeff Barrett, W.B.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.L.; Volz, E. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 25 March 2021).
- Robson, F.; Khan, K.S.; Le, T.K.; Paris, C.; Demirbag, S.; Barfuss, P.; Rocchi, P.; Ng, W.L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79, 710–727. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.K.; Huang, I.C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [Green Version]
- Song, H.D.; Tu, C.C.; Zhang, G.W.; Wang, S.Y.; Zheng, K.; Lei, L.C.; Chen, Q.X.; Gao, Y.W.; Zhou, H.Q.; Xiang, H.; et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. USA 2005, 102, 2430–2435. [Google Scholar] [CrossRef] [Green Version]
- Singh, J.; Samal, J.; Kumar, V.; Sharma, J.; Agrawal, U.; Ehtesham, N.Z.; Sundar, D.; Rahman, S.A.; Hira, S.; Hasnain, S.E. Structure-Function Analyses of New SARS-CoV-2 Variants B.1.1.7, B.1.351 and B.1.1.28.1: Clinical, Diagnostic, Therapeutic and Public Health Implications. Viruses 2021, 13, 439. [Google Scholar] [CrossRef] [PubMed]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Volz, E.; Hill, V.; McCrone, J.T.; Price, A.; Jorgensen, D.; O’Toole, Á.; Southgate, J.; Johnson, R.; Jackson, B.; Nascimento, F.F.; et al. Evaluating the Effects of SARS-CoV-2 Spike Mutation D614G on Transmissibility and Pathogenicity. Cell 2021, 184, 64–75.e11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jackson, C.B.; Mou, H.; Ojha, A.; Peng, H.; Quinlan, B.D.; Rangarajan, E.S.; Pan, A.; Vanderheiden, A.; Suthar, M.S.; et al. SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nat. Commun. 2020, 11, 6013. [Google Scholar] [CrossRef]
- Isabel, S.; Graña-Miraglia, L.; Gutierrez, J.M.; Bundalovic-Torma, C.; Groves, H.E.; Isabel, M.R.; Eshaghi, A.; Patel, S.N.; Gubbay, J.B.; Poutanen, T.; et al. Evolutionary and structural analyses of SARS-CoV-2 D614G spike protein mutation now documented worldwide. Sci. Rep. 2020, 10, 14031. [Google Scholar] [CrossRef]
- Deng, X.; Gu, W.; Federman, S.; Plessis, L.D.; Pybus, O.G.; Faria, N.R.; Wang, C.; Yu, G.; Bushnell, B.; Pan, C.Y.; et al. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science 2020, 369, 582–587. [Google Scholar] [CrossRef]
- Cai, Y.; Zhang, J.; Xiao, T.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rawson, S.; Rits-Volloch, S.; Chen, B. Distinct conformational states of SARS-CoV-2 spike protein. Science 2020, 369, 1586–1592. [Google Scholar] [CrossRef]
- Rozzi, R.; Armesto, J.J.; Gutiérrez, J.R.; Massardo, F.; Likens, G.E.; Anderson, C.B.; Poole, A.; Moses, K.P.; Hargrove, E.; Mansilla, A.O.; et al. Integrating Ecology and Environmental Ethics: Earth Stewardship in the Southern End of the Americas. BioScience 2012, 62, 226–236. [Google Scholar] [CrossRef] [Green Version]
- Castillo, A.E.; Parra, B.; Tapia, P.; Acevedo, A.; Lagos, J.; Andrade, W.; Arata, L.; Leal, G.; Barra, G.; Tambley, C.; et al. Phylogenetic analysis of the first four SARS-CoV-2 cases in Chile. J. Med. Virol. 2020, 92, 1562–1566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artic Network. Available online: https://artic.network/ (accessed on 25 March 2021).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Liu, K.; Zhang, H.; Zhang, L.; Bian, Y.; Huang, L. CoV-Seq, a New Tool for SARS-CoV-2 Genome Analysis and Visualization: Development and Usability Study. J. Med. Internet Res. 2020, 22, e22299. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Machado, M.R.; Pantano, S. SIRAH tools: Mapping, backmapping and visualization of coarse-grained models. Bioinformatics 2016, 32, 1568–1570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. In Solving Software Challenges for Exascale; Markidis, S., Laure, E., Eds.; Lecture Notes in Computer Science; Springer International Publishing: Cham, Switzerland, 2015; pp. 3–27. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Darré, L.; Machado, M.R.; Dans, P.D.; Herrera, F.E.; Pantano, S. Another coarse grain model for aqueous solvation: WAT FOUR? J. Chem. Theory Comput. 2010, 6, 3793–3807. [Google Scholar] [CrossRef]
- Machado, M.R.; Pantano, S. Split the charge difference in two! A rule of thumb for adding proper amounts of ions in MD simulations. J. Chem. Theory Comput. 2020, 16, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef]
- Schrödinger, L.; DeLano, W. PyMOL. 2020. Available online: http://www.pymol.org/pymol (accessed on 25 March 2021).
- Community, B.O. Blender-a 3D Modelling and Rendering Package. 2021. Available online: http://www.blender.org (accessed on 25 March 2021).
- González-Puelma, J.; Aldridge, J.; Montes de Oca, M.; Pinto, M.; Uribe-Paredes, R.; Fernández-Goycoolea, J.; Alvarez Saravia, D.; Álvarez, H.; Encina, G.; Weitzel, T.; et al. SIRAH-CoV2 Initiative: T307I Mutation in a SARS-CoV-2 Spike from Sub-Antarctic Chile. 2021. Available online: https://zenodo.org/record/4730896#.YJuTDKERWUk (accessed on 28 April 2021). [CrossRef]
- Garay, P.G.; Barrera, E.E.; Klein, F.; Machado, M.R.; Soñora, M.; Pantano, S. The SIRAH-CoV-2 Initiative: A coarse-grained simulations’ dataset of the sars-cov-2 proteome. Front. Med. Technol. 2021, 3, 4. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Johnson, B.A.; Xie, X.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; Zhang, L.; et al. Furin Cleavage Site Is Key to SARS-CoV-2 Pathogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Gobeil, S.M.C.; Janowska, K.; McDowell, S.; Mansouri, K.; Manne, K.; Stalls, V.; Kopp, M.; Henderson, R.; Edwards, R.J.; Haynes, B.F.; et al. D614G mutation alters SARS-CoV-2 spike conformational dynamics and protease cleavage susceptibility at the S1/S2 junction. bioRxiv 2020, 34. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020, 183, 739–751.e8. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Moreira, R.A.; Chwastyk, M.; Baker, J.L.; Guzman, H.V.; Poma, A.B. Quantitative determination of mechanical stability in the novel coronavirus spike protein. Nanoscale 2020, 12, 16409–16413. [Google Scholar] [CrossRef]
- Moreira, R.A.; Guzman, H.V.; Boopathi, S.; Baker, J.L.; Poma, A.B. Characterization of structural and energetic differences between conformations of the SARS-CoV-2 spike protein. Materials 2020, 13, 5362. [Google Scholar] [CrossRef]
- Barros, E.P.; Casalino, L.; Gaieb, Z.; Dommer, A.C.; Wang, Y.; Fallon, L.; Raguette, L.; Belfon, K.; Simmerling, C.; Amaro, R.E. The flexibility of ACE2 in the context of SARS-CoV-2 infection. Biophys. J. 2021, 120, 1072–1084. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, Y.; Xiao, T.; Lu, J.; Peng, H.; Sterling, S.M.; Walsh, R.M.; Rits-Volloch, S.; Zhu, H.; Woosley, A.N.; et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science 2021, 372, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Zheng, Y.; Yang, Y.; Liu, C.; Geng, Q.; Luo, C.; Zhang, W.; Li, F. Cryo-EM structure of infectious bronchitis coronavirus spike protein reveals structural and functional evolution of coronavirus spike proteins. PLoS Pathog. 2018, 14, e1007009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, T.; Tsybovsky, Y.; Gorman, J.; Rapp, M.; Cerutti, G.; Chuang, G.Y.; Katsamba, P.S.; Sampson, J.M.; Schön, A.; Bimela, J.; et al. Cryo-EM structures of SARS-CoV-2 spike without and with ACE2 reveal a pH-dependent switch to mediate endosomal positioning of receptor-binding domains. Cell Host Microbe 2020, 28, 867–879. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making Sense of Mutation: What D614G Means for the COVID-19 Pandemic Remains Unclear. Cell 2020, 182, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Altman, M.O.; Angeletti, D.; Yewdell, J.W. Antibody Immunodominance: The Key to Understanding Influenza Virus Antigenic Drift. Viral Immunol. 2018, 31, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Diehl, W.E.; Lin, A.E.; Grubaugh, N.D.; Carvalho, L.M.; Kim, K.; Kyawe, P.P.; McCauley, S.M.; Donnard, E.; Kucukural, A.; McDonel, P.; et al. Ebola Virus Glycoprotein with Increased Infectivity Dominated the 2013–2016 Epidemic. Cell 2016, 167, 1088–1098.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zehender, G.; Lai, A.; Bergna, A.; Meroni, L.; Riva, A.; Balotta, C.; Tarkowski, M.; Gabrieli, A.; Bernacchia, D.; Rusconi, S.; et al. Genomic characterization and phylogenetic analysis of SARS-COV-2 in Italy. J. Med Virol. 2020, 92, 1637–1640. [Google Scholar] [CrossRef] [Green Version]
- Disease Prevention, E.C. Rapid increase of a SARS-CoV-2 variant with multiple spike protein mutations observed in the United Kingdom—20 December 2020. ECDC Stockh. 2020. [Google Scholar]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Vuong, W.; Khan, M.B.; Fischer, C.; Arutyunova, E.; Lamer, T.; Shields, J.; Saffran, H.A.; McKay, R.T.; van Belkum, M.J.; Joyce, M.A.; et al. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. [Google Scholar] [CrossRef]
- Leary, S.; Gaudieri, S.; Chopra, A.; Pakala, S.; Alves, E.; John, M.; Das, S.; Mallal, S.; Phillips, E. Three adjacent nucleotide changes spanning two residues in SARS-CoV-2 nucleoprotein: Possible homologous recombination from the transcription-regulating sequence. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Berger, I.; Schaffitzel, C. The SARS-CoV-2 spike protein: Balancing stability and infectivity. Cell Res. 2020, 30, 1059–1060. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Henderson, R.; Edwards, R.J.; Mansouri, K.; Janowska, K.; Stalls, V.; Gobeil, S.M.; Kopp, M.; Li, D.; Parks, R.; Hsu, A.L.; et al. Controlling the SARS-CoV-2 spike glycoprotein conformation. Nat. Struct. Mol. Biol. 2020, 27, 925–933. [Google Scholar] [CrossRef]
- Ozer, E.A.; Simons, L.M.; Adewumi, O.M.; Fowotade, A.A.; Omoruyi, E.C.; Adeniji, J.A.; Dean, T.J.; Taiwo, B.O.; Hultquist, J.F.; Lorenzo-Redondo, R. High prevalence of SARS-CoV-2 B. 1.1. 7 (UK variant) and the novel B. 1.5. 2.5 lineage in Oyo State, Nigeria. medRxiv. [CrossRef]
- Kemp, S.A.; Meng, B.; Ferriera, I.A.; Datir, R.; Harvey, W.T.; Papa, G.; Lytras, S.; Collier, D.A.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence and transmission of a SARS-CoV-2 spike deletion H69/V70. bioRxiv 2021. [Google Scholar] [CrossRef]
- Maggi, F.; Novazzi, F.; Genoni, A.; Baj, A.; Spezia, P.G.; Focosi, D.; Zago, C.; Colombo, A.; Cassani, G.; Pasciuta, R.; et al. 1 in Traveler Returning from Brazil to Italy. Emerg. Infect. Dis. 2021, 27, 1249. [Google Scholar] [CrossRef] [PubMed]
- Yadav, P.D.; Sapkal, G.N.; Abraham, P.; Ella, R.; Deshpande, G.; Patil, D.Y.; Nyayanit, D.A.; Gupta, N.; Sahay, R.R.; Shete, A.M.; et al. 1.617 with sera of BBV152 vaccinees. bioRxiv 2021. [Google Scholar] [CrossRef]
- Peacock, T.P.; Penrice-Randal, R.; Hiscox, J.A.; Barclay, W.S. SARS-CoV-2 one year on: Evidence for ongoing viral adaptation. J. Gen. Virol. 2021, 102, 001584. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
González-Puelma, J.; Aldridge, J.; Montes de Oca, M.; Pinto, M.; Uribe-Paredes, R.; Fernández-Goycoolea, J.; Alvarez-Saravia, D.; Álvarez, H.; Encina, G.; Weitzel, T.; et al. Mutation in a SARS-CoV-2 Haplotype from Sub-Antarctic Chile Reveals New Insights into the Spike’s Dynamics. Viruses 2021, 13, 883. https://doi.org/10.3390/v13050883
González-Puelma J, Aldridge J, Montes de Oca M, Pinto M, Uribe-Paredes R, Fernández-Goycoolea J, Alvarez-Saravia D, Álvarez H, Encina G, Weitzel T, et al. Mutation in a SARS-CoV-2 Haplotype from Sub-Antarctic Chile Reveals New Insights into the Spike’s Dynamics. Viruses. 2021; 13(5):883. https://doi.org/10.3390/v13050883
Chicago/Turabian StyleGonzález-Puelma, Jorge, Jacqueline Aldridge, Marco Montes de Oca, Mónica Pinto, Roberto Uribe-Paredes, José Fernández-Goycoolea, Diego Alvarez-Saravia, Hermy Álvarez, Gonzalo Encina, Thomas Weitzel, and et al. 2021. "Mutation in a SARS-CoV-2 Haplotype from Sub-Antarctic Chile Reveals New Insights into the Spike’s Dynamics" Viruses 13, no. 5: 883. https://doi.org/10.3390/v13050883
APA StyleGonzález-Puelma, J., Aldridge, J., Montes de Oca, M., Pinto, M., Uribe-Paredes, R., Fernández-Goycoolea, J., Alvarez-Saravia, D., Álvarez, H., Encina, G., Weitzel, T., Muñoz, R., Olivera-Nappa, Á., Pantano, S., & Navarrete, M. A. (2021). Mutation in a SARS-CoV-2 Haplotype from Sub-Antarctic Chile Reveals New Insights into the Spike’s Dynamics. Viruses, 13(5), 883. https://doi.org/10.3390/v13050883