
An Early Block in the Replication of the Atypical Bluetongue Virus Serotype 26 in Culicoides Cells Is Determined by Its Capsid Proteins

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses and Cells
2.2. Time-Course Experiments
2.3. Virus Titration
2.4. Virus Purification
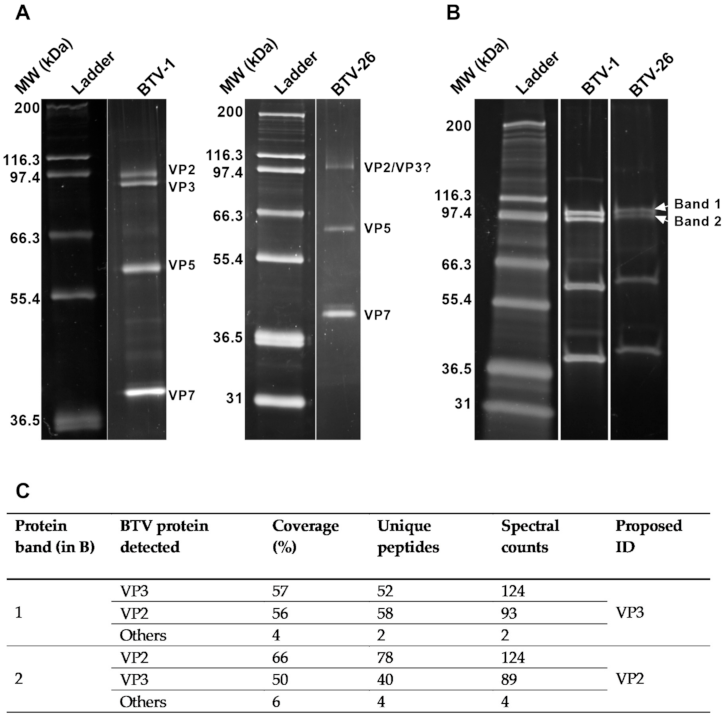
2.5. Protein Identification by Mass Spectrometry
2.6. Fluorescent Microscopy
2.7. Binding Assay
2.8. Transmission Electron Microscopy (TEM)
2.9. Oral Infection of Adult Culicoides sonorensis Midges
2.10. Statistical Analysis
3. Results
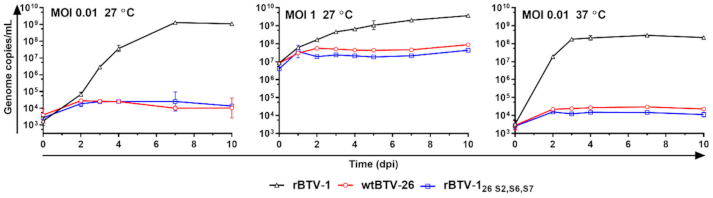
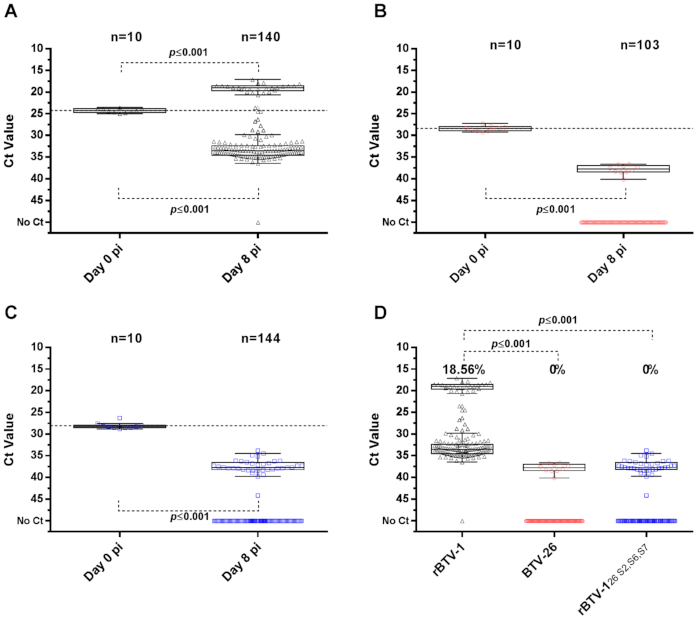
3.1. BTV-26 KUW2010/02 Is Unable to Replicate in Arthropod-Derived Cells In Vitro
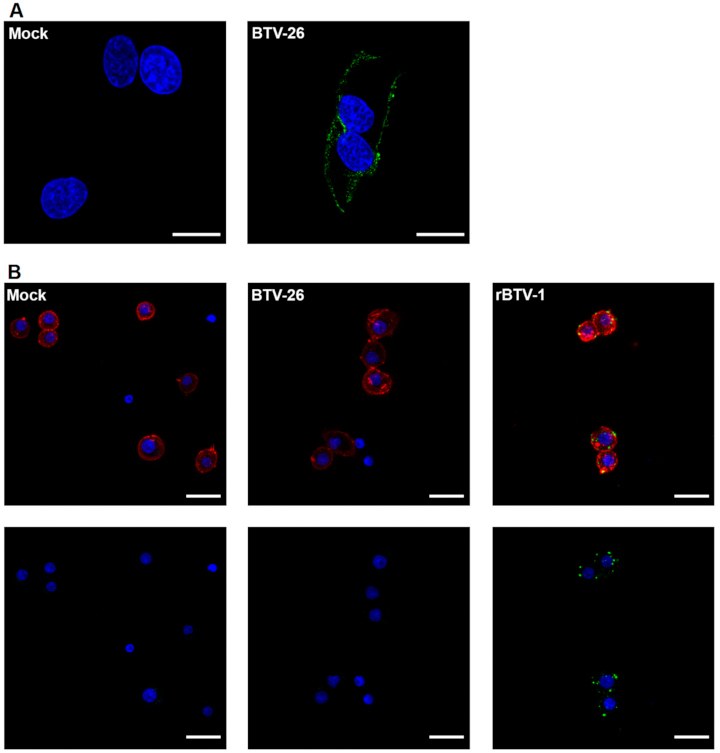
3.2. Cell Binding Studies of Purified BTV-26 in Culicoides-Derived Cells
3.3. The Ability of the BTV-26 Capsid to Block Virus Replication in KC Cells
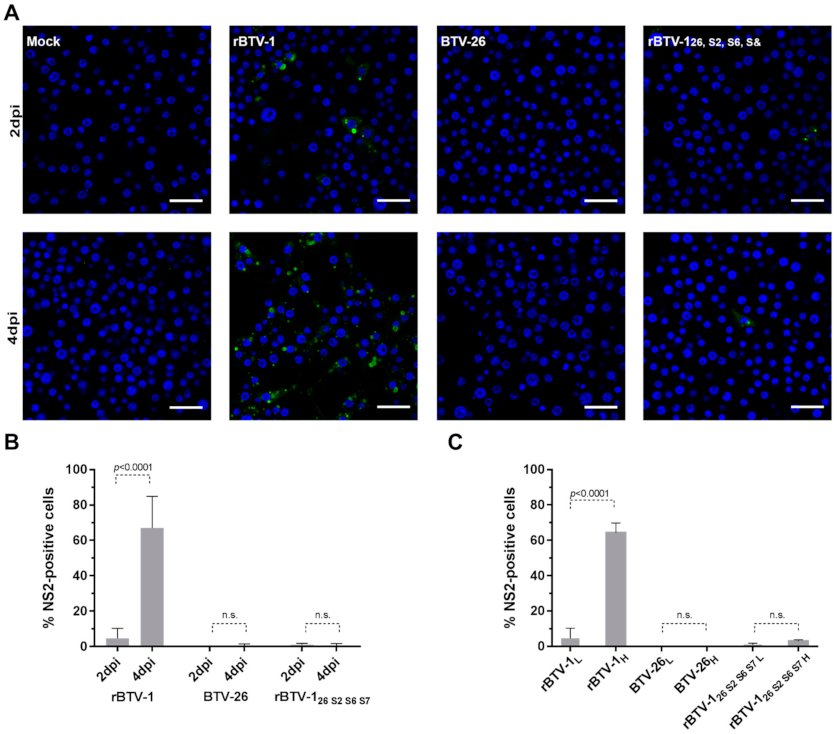
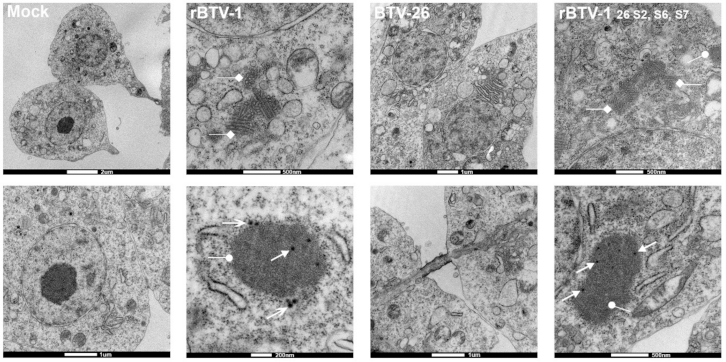
3.4. The Capsid of BTV-26 Blocks the Early Stages of Virus Replication Cycle in KC Cells
3.5. Capsid Proteins of BTV-26 Restrict BTV Replication in Adult C. sonorensis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Alexander, K.A.; MacLachlan, N.J.; Kat, P.W.; House, C.; O’Brien, S.J.; Lerche, N.W.; Sawyer, M.; Frank, L.G.; Holekamp, K.; Smale, L.; et al. Evidence of natural bluetongue virus infection among African carnivores. Am. J. Trop. Med. Hyg. 1994, 51, 568–576. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jauniaux, T.P.; De Clercq, K.E.; Cassart, D.E.; Kennedy, S.; Vandenbussche, F.E.; Vandemeulebroucke, E.L.; Vanbinst, T.M.; Verheyden, B.I.; Goris, N.E.; Coignoul, F.L. Bluetongue in Eurasian lynx. Emerg. Infect. Dis. 2008, 14, 1496–1498. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.; Lacroux, C.; Leger, S.; Top, S.; Goyeau, K.; Deplanche, M.; Lemaire, M. Lethal bluetongue virus serotype 1 infection in llamas. Emerg. Infect. Dis. 2009, 15, 608–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Organisation for Animal Health (OIE): Paris, France. Available online: https://www.oie.int/en/what-we-do/animal-health-and-welfare/animal-diseases/?/ (accessed on 14 May 2021).
- Hewat, E.H.; Booth, T.F.; Roy, P. Structure of bluetongue virus particles by cryoelectron microscopy. J. Struct. Biol. 1992, 109, 61–69. [Google Scholar] [CrossRef]
- Mertens, P.P.C.; Maan, S.; Samuel, A.; Attoui, H. Orbivirus, Reoviridae; Virus Taxonomy VIIIth Report of the ICTVC; Fauquet, M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., Eds.; Elsevier/Academic Press: London, UK, 2004. [Google Scholar]
- Maan, S.; Maan, N.S.; Samuel, A.R.; Rao, S.; Attoui, H.; Mertens, P.P.C. Analysis and phylogenetic comparisons of full-length VP2 genes of the 24 bluetongue virus serotypes. J. Gen. Virol. 2007, 88, 621–630. [Google Scholar] [CrossRef]
- Gibbs, E.P.J.; Lawman, M.J.P.; Herniman, K.A.J. Preliminary observations on transplacental infection of bluetongue virus in sheep—Possible overwintering mechanism. Res. Vet. Sci. 1979, 27, 118–120. [Google Scholar] [CrossRef]
- Menzies, F.D.; McCullough, S.J.; McKeown, I.M.; Forster, J.L.; Jess, S.; Batten, C.; Murchie, A.K.; Gloster, J.; Fallows, J.G.; Pelgrim, W.; et al. Evidence for transplacental and contact transmission of bluetongue virus in cattle. Vet. Rec. 2008, 163, 203–209. [Google Scholar] [CrossRef] [Green Version]
- Darpel, K.E.; Batten, C.A.; Veronesi, E.; Williamson, S.; Anderson, P.; Dennison, M.; Clifford, S.; Smith, C.; Philips, L.; Bidewell, C.; et al. Transplacental transmission of bluetongue virus 8 in cattle, UK. Emerg. Infect. Dis. 2009, 15, 2025–2028. [Google Scholar] [CrossRef]
- Borkent, A. The pupae of the biting midges of the world (Diptera: Ceratopogonidae), with a generic key and analysis of the phylogenetic relationships between genera. Zootaxa 2014, 3879, 1–327. [Google Scholar] [CrossRef] [Green Version]
- Purse, B.V.; Carpenter, S.; Venter, G.J.; Bellis, G.; Mullens, B.A. Bionomics of temperate and tropical Culicoides midges: Knowledge gaps and consequences for transmission of Culicoides-borne viruses. Annu. Rev. Entomol. 2015, 60, 373–392. [Google Scholar] [CrossRef]
- Purse, B.V.; Mellor, P.S.; Rogers, D.J.; Samuel, A.R.; Mertens, P.P.C.; Baylis, M. Climate change and the recent emergence of bluetongue in Europe. Nat. Rev. Microbiol. 2005, 3, 171–181. [Google Scholar] [CrossRef]
- Mellor, P.S.; Carpenter, S.; Harrup, L.; Baylis, M.; Mertens, P.P.C. Bluetongue in Europe and the Mediterranean Basin: History of occurrence prior to 2006. Prev. Vet. Med. 2008, 87, 4–20. [Google Scholar] [CrossRef]
- Curini, V.; Marcacci, M.; Tonelli, A.; Di Teodoro, G.; Di Domenico, M.; D’Alterio, N.; Portanti, O.; Ancora, M.; Savini, G.; Panfili, M.; et al. Molecular typing of Bluetongue virus using the nCounter® analysis system platform. J. Virol. Methods 2019, 269, 64–69. [Google Scholar] [CrossRef]
- Ries, C.; Sharav, T.; Tseren-Ochir, E.O.; Beer, M.; Hoffmann, B. Putative novel serotypes ‘33’ and ‘35’ in clinically healthy small ruminants in Mongolia expand the group of atypical BTV. Viruses 2020, 13, 42. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Renzullo, S.; Mader, M.; Chaignat, V.; Worwa, G.; Thuer, B. Genetic characterization of toggenburg orbivirus, a new bluetongue virus, from goats, Switzerland. Emerg. Infect. Dis. 2008, 14, 1855–1861. [Google Scholar] [CrossRef]
- Batten, C.; Darpel, K.; Henstock, M.; Fay, P.; Veronesi, E.; Gubbins, S.; Graves, S.; Frost, L.; Oura, C. Evidence for Transmission of Bluetongue virus serotype 26 through direct contact. PLoS ONE 2014, 9, e96049. [Google Scholar] [CrossRef] [Green Version]
- Pullinger, G.D.; Guimera Busquets, M.; Nomikou, K.; Boyce, M.; Attoui, H.; Mertens, P.P. Identification of the genome segments of Bluetongue virus serotype 26 (Isolate KUW2010/02) that restrict replication in a Culicoides sonorensis cell line (KC cells). PLoS ONE 2016, 11, e0149709. [Google Scholar] [CrossRef]
- Breard, E.; Schulz, C.; Sailleau, C.; Bernelin-Cottet, C.; Viarouge, C.; Vitour, D.; Guillaume, B.; Caignard, G.; Gorlier, A.; Attoui, H.; et al. Bluetongue virus serotype 27: Experimental infection of goats, sheep and cattle with three BTV-27 variants reveal atypical characteristics and likely direct contact transmission BTV-27 between goats. Transbound. Emerg. Dis. 2018, 65, e251–e263. [Google Scholar] [CrossRef]
- Lorusso, A.; Baba, D.; Spedicato, M.; Teodori, L.; Bonfini, B.; Marcacci, M.; Di Provvido, A.; Isselmou, K.; Marini, V.; Carmine, I.; et al. Bluetongue virus surveillance in the Islamic Republic of Mauritania: Is serotype 26 circulating among cattle and dromedaries? Infect. Genet. Evol. 2016, 40, 109–112. [Google Scholar] [CrossRef]
- Jennings, D.M.; Mellor, P.S. Variation in the responses of Culicoides variipennis (Diptera, Ceratopogonidae) to oral infection with Bluetongue virus. Arch. Virol. 1987, 95, 177–182. [Google Scholar] [CrossRef]
- Tabachnick, W.J. Genetic control of oral susceptibility to infection of Culicoides variipennis with Bluetongue virus. Am. J. Trop. Med. Hyg. 1991, 45, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.; Leake, C.J.; Mertens, P.P.C.; Mellor, P.S. The barriers to bluetongue virus infection, dissemination and transmission in the vector, Culicoides variipennis (Diptera: Ceratopogonidae). Arch. Virol. 1999, 144, 747–761. [Google Scholar] [CrossRef] [PubMed]
- Veronesi, E.; Antony, F.; Gubbins, S.; Golding, N.; Blackwell, A.; Mertens, P.P.C.; Brownlie, J.; Darpel, K.E.; Mellor, P.S.; Carpenter, S. Measurement of the infection and dissemination of Bluetongue virus in Culicoides biting midges using a semi-quantitative RT-PCR assay and isolation of infectious virus. PLoS ONE 2013, 8, e70800. [Google Scholar] [CrossRef] [PubMed]
- Tabachnick, W.J. Nature, nurture and evolution of intra-species variation in mosquito arbovirus transmission competence. Int. J. Environ. Res. Public Health 2013, 10, 249–277. [Google Scholar] [CrossRef] [Green Version]
- Severson, D.W.; Behura, S.K. Genome investigations of vector competence in aedes aegypti to inform novel arbovirus disease control approaches. Insects 2016, 7, 58. [Google Scholar] [CrossRef]
- Mills, M.K.; Michel, K.; Pfannenstiel, R.S.; Ruder, M.G.; Veronesi, E.; Nayduch, D. Culicoides-Virus interactions: Infection barriers and possible factors underlying vector competence. Curr. Opin. Insect. Sci. 2017, 22, 7–15. [Google Scholar] [CrossRef]
- Feenstra, F.; Drolet, B.S.; Boonstra, J.; van Rijn, P.A. Non-Structural protein NS3/NS3a is required for propagation of bluetongue virus in Culicoides sonorensis. Parasites Vectors 2015, 8, 476. [Google Scholar] [CrossRef] [Green Version]
- Belhouchet, M.; Mohd Jaafar, F.; Firth, A.E.; Grimes, J.M.; Mertens, P.P.; Attoui, H. Detection of a fourth orbivirus non-structural protein. PLoS ONE 2011, 6, e25697. [Google Scholar] [CrossRef] [Green Version]
- Ratinier, M.; Caporale, M.; Golder, M.; Franzoni, G.; Allan, K.; Nunes, S.F.; Armezzani, A.; Bayoumy, A.; Rixon, F.; Shaw, A.; et al. Identification and characterization of a novel non-structural protein of bluetongue virus. PLoS Pathog. 2011, 7, e1002477. [Google Scholar] [CrossRef] [Green Version]
- Stewart, M.; Hardy, A.; Barry, G.; Pinto, R.M.; Caporale, M.; Melzi, E.; Hughes, J.; Taggart, A.; Janowicz, A.; Varela, M.; et al. Characterization of a second open reading frame in genome segment 10 of bluetongue virus. J. Gen. Virol. 2015, 96, 3280–3293. [Google Scholar] [CrossRef]
- Stuart, D.I.; Gouet, P.; Grimes, J.M.; Malby, R.; Diprose, J.M.; Zientara, S.; Burroughs, J.N.; Mertens, P.P.C. Structual studies of orbivirus particles. Arch. Virol. 1998, 14, 235–250. [Google Scholar]
- Grimes, J.M.; Burroughs, J.N.; Gouet, P.; Diprose, J.M.; Malby, R.; Ziéntara, S.; Mertens, P.P.C.; Stuart, D.I. The atomic structure of the bluetongue virus core. Nature 1998, 395, 470–478. [Google Scholar] [CrossRef]
- Forzan, M.; Marsh, M.; Roy, P. Bluetongue virus entry into cells. J. Virol. 2007, 81, 4819–4827. [Google Scholar] [CrossRef] [Green Version]
- Gold, S.; Monaghan, P.; Mertens, P.; Jackson, T. A clathrin independent macropinocytosis-like entry mechanism used by bluetongue virus-1 during infection of BHK cells. PLoS ONE 2010, 5, e11360. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Patel, A.; Celma, C.C.; Yu, X.; Roy, P.; Zhou, Z.H. Atomic model of a nonenveloped virus reveals pH sensors for a coordinated process of cell entry. Nat. Struct. Mol. Biol. 2016, 23, 74–80. [Google Scholar] [CrossRef]
- Stevens, L.M.; Moffat, K.; Cooke, L.; Nomikou, K.; Mertens, P.P.C.; Jackson, T.; Darpel, K.E. A low-passage insect-cell isolate of bluetongue virus uses a macropinocytosis-like entry pathway to infect natural target cells derived from the bovine host. J. Gen. Virol. 2019, 100, 568–582. [Google Scholar] [CrossRef]
- Mertens, P.P.C.; Burroughs, J.N.; Walton, A.; Wellby, M.P.; Fu, H.; O’Hara, R.S.; Brookes, S.M.; Mellor, P.S. Enhanced infectivity of modified Bluetongue virus particles for two insect cell lines and for two Culicoides vector species. Virology 1996, 217, 582–593. [Google Scholar] [CrossRef] [Green Version]
- Darpel, K.E.; Langner, K.F.A.; Nimtz, M.; Anthony, S.J.; Brownlie, J.; Takamatsu, H.-H.; Mellor, P.S.; Mertens, P.P.C. Saliva proteins of vector Culicoides modify structure and infectivity of bluetongue virus particles. PLoS ONE 2011, 6, e17545. [Google Scholar] [CrossRef]
- Maan, S.; Maan, N.S.; Nomikou, K.; Veronesi, E.; Bachanek-Bankowska, K.; Belaganahalli, M.N.; Attoui, H.; Mertens, P.P. Complete genome characterisation of a novel 26th bluetongue virus serotype from Kuwait. PLoS ONE 2011, 6, e26147. [Google Scholar] [CrossRef] [Green Version]
- Schulz, C.; Breard, E.; Sailleau, C.; Jenckel, M.; Viarouge, C.; Vitour, D.; Palmarini, M.; Gallois, M.; Hoper, D.; Hoffmann, B.; et al. Bluetongue virus serotype 27: Detection and characterization of two novel variants in Corsica, France. J. Gen. Virol. 2016, 97, 2073–2083. [Google Scholar] [CrossRef]
- Bumbarov, V.; Golender, N.; Jenckel, M.; Wernike, K.; Beer, M.; Khinich, E.; Zalesky, O.; Erster, O. Characterization of bluetongue virus serotype 28. Transbound. Emerg. Dis. 2020, 67, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Gu, W.; Li, Z.; Zhang, L.; Liao, D.; Song, J.; Baoxin, S.; Hasimu, J.; Li, Z.; Yang, Z.; et al. Novel putative Bluetongue virus serotype 29 isolated from inapparently infected goat in Xinjiang of China. Transbound. Emerg. Dis. 2021, 1–13. [Google Scholar] [CrossRef]
- Sato, M.; Maeda, N.; Yoshida, H.; Urade, M.; Saito, S. Plaque formation of herpes virus hominis type 2 and rubella virus in variants isolated from the colonies of BHK21/WI-2 cells formed in soft agar. Arch. Virol. 1977, 53, 269–273. [Google Scholar] [CrossRef]
- Wechsler, S.J.; McHolland, L.E.; Tabachnick, W.J. Cell-Lines from Culicoides variipennis (Diptera, Ceratopogonidae) support replication of Bluetongue virus. J. Invertebr. Pathol. 1989, 54, 385–393. [Google Scholar] [CrossRef]
- Miller, M.L.; Brown, D.T. Morphogenesis of Sindbis virus in three subclones of Aedes albopictus (mosquito) cells. J. Virol. 1992, 66, 4180–4190. [Google Scholar] [CrossRef] [Green Version]
- Kurtti, T.J.; Munderloh, U.G.; Samish, M. Effect of medium supplements on tick cells in culture. J. Parasitol. 1982, 68, 930–935. [Google Scholar] [CrossRef]
- Bell-Sakyi, L. Continuous cell lines from the tick Hyalomma anatolicum anatolicum. J. Parasitol. 1991, 77, 1006–1008. [Google Scholar] [CrossRef]
- Maan, N.S.; Maan, S.; Belaganahalli, M.; Pullinger, G.; Montes, A.J.A.; Gasparini, M.R.; Guimera, M.; Nomikou, K.; Mertens, P.P.C. A quantitative real-time reverse transcription PCR (qRT-PCR) assay to detect genome segment 9 of all 26 bluetongue virus serotypes. J. Virol. Methods 2015, 213, 118–126. [Google Scholar] [CrossRef]
- Kärber, G. Beitrag zur kollektiven Behandlung pharmakologischer Reihenversuche [A contribution to the collective treatment of a pharmacological experimental series]. Arch. Exp. Pathol. Pharmakol. 1931, 162, 480–483. [Google Scholar] [CrossRef]
- Mertens, P.P.C.; Burroughs, J.N.; Anderson, J. Purification and properties of virus-particles, infectious subviral particles, and cores of Bluetongue virus serotype-1 and serotype-4. Virology 1987, 157, 375–386. [Google Scholar] [CrossRef]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-Gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef] [PubMed]
- Aljabr, W.; Armstrong, S.; Rickett, N.Y.; Pollakis, G.; Touzelet, O.; Cloutman-Green, E.; Matthews, D.A.; Hiscox, J.A. High resolution analysis of respiratory syncytial virus infection in vivo. Viruses 2019, 11, 926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, B.; Zhang, K.; Hendrie, C.; Liang, C.; Li, M.; Doherty-Kirby, A.; Lajoie, G. PEAKS: Powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid. Commun. Mass. Spectrom. 2003, 17, 2337–2342. [Google Scholar] [CrossRef] [PubMed]
- Boorman, J. The maintenance of laboratory colonies of Culicoides variipennis (Coq), C. nubeculosus (Mg) and C.riethi Kieff. (Diptera, Ceratopogonidae). Bull. Entomol. Res. 1974, 64, 371–377. [Google Scholar] [CrossRef]
- Veronesi, E.; Mertens, P.P.C.; Shaw, A.E.; Brownlie, J.; Mellor, P.S.; Carpenter, S.T. Quantifying bluetongue virus in adult Culicoides biting midges (Diptera: Ceratopogonidae). J. Med Entomol. 2008, 45, 129–132. [Google Scholar] [CrossRef] [Green Version]
- Batten, C.A.; Henstock, M.R.; Bin-Tarif, A.; Steedman, H.M.; Waddington, S.; Edwards, L.; Oura, C.A. Bluetongue virus serotype 26: Infection kinetics and pathogenesis in Dorset Poll sheep. Vet. Microbiol. 2012, 157, 119–124. [Google Scholar] [CrossRef]
- Patel, A.; Roy, P. The molecular biology of Bluetongue virus replication. Virus Res. 2014, 182, 5–20. [Google Scholar] [CrossRef]
- Brookes, S.M.; Hyatt, A.D.; Eaton, B.T. Characterization of virus inclusion bodies in bluetongue virus-infected cells. J. Gen. Virol. 1993, 74, 525–530. [Google Scholar] [CrossRef]
- Labadie, T.; Roy, P. A non-enveloped arbovirus released in lysosome-derived extracellular vesicles induces super-infection exclusion. PLoS Pathog. 2020, 16, e1009015. [Google Scholar] [CrossRef]
- Gould, A.R.; Hyatt, A.D.; Eaton, B.T. Morphogenesis of a bluetongue virus variant with an amino acid alteration at a neutralization site in the outer coat protein, VP2. Virology 1988, 165, 23–32. [Google Scholar] [CrossRef]
- Beaton, A.R.; Rodriguez, J.; Reddy, Y.K.; Roy, P. The membrane trafficking protein calpactin forms a complex with bluetongue virus protein NS3 and mediates virus release. Proc. Natl. Acad. Sci. USA 2002, 99, 13154–13159. [Google Scholar] [CrossRef] [Green Version]
- Mertens, P.P.; Diprose, J.; Maan, S.; Singh, K.P.; Attoui, H.; Samuel, A.R. Bluetongue virus replication, molecular and structural biology. Vet. Ital. 2004, 40, 426–437. [Google Scholar]
- Bhattacharya, B.; Noad, R.J.; Roy, P. Interaction between Bluetongue virus outer capsid protein VP2 and vimentin is necessary for virus egress. Virol. J. 2007, 15, 4–7. [Google Scholar]
- Reber, A.; Kreienbrock, L.; Casati, S.; Chaignat, V.; Schwermer, H. Putative risk factors for infections with Toggenburg Orbivirus in goat herds in Southern Switzerland (Canton of Ticino). Vet. Microbiol. 2012, 160, 29–34. [Google Scholar] [CrossRef]
- Zhang, X.; Boyce, M.; Bhattacharya, B.; Zhang, X.; Schein, S.; Roy, P.; Zhou, Z.H. Bluetongue virus coat protein VP2 contains sialic acid-binding domains, and VP5 resembles enveloped virus fusion proteins. Proc. Natl. Acad. Sci. USA 2010, 107, 6292–6297. [Google Scholar] [CrossRef] [Green Version]
- Feenstra, F.; Pap, J.S.; van Rijn, P.A. Application of bluetongue Disabled Infectious Single Animal (DISA) vaccine for different serotypes by VP2 exchange or incorporation of chimeric VP2. Vaccine 2015, 33, 812–818. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Virus (ORC number) 1 | Cell Passage History 2 | Origin 3 | Comments |
|---|---|---|---|
| BTV-1 (RSArrrr/01) | E2, BHK9, BSR1 * | Natural (TPI) | Strain used for segment cloning, identified as wtBTV-1 |
| BTV-26 (KUW2010/02) | E1, BHK2, BSR1 | Natural (TPI) | Strain used for segment cloning and infections, identified as BTV-26 |
| rBTV-1 (BTV-RV0023) | BSR3 | Synthetic | Rescued strain, derived from BTV-1 RSArrrr/01 cloned segments |
| rBTV-126 S2,S6,S7 (BTV-RV0020) | BSR3 | Synthetic | Rescued strain comprised of wtBTV-1 derived backbone, with genome-segments 2, 6 and 7 derived from BTV-26 |
| Cell Line | No. of Seeded Cells | Incubation (°C) | Maintenance Medium |
|---|---|---|---|
| BSR | 1 × 105 cells/well (24 well plate) 3 × 106 cells/flask (T25 cm2 flask) | 37 ± 1 with 5% CO2 | DMEM GlutaMAX™ + 1% HI-FBS + 1% Pen/Strep |
| BFA | 1 × 105 cells/well (24 well plate) | 37 ± 1 with 5% CO2 | Nutrient Mixture F-12 Ham’s medium + 1% HI-FBS + 1% Pen/Strep |
| KC | 7 × 105 cells/well (24 well plate) 2.5 × 107 cells/flask (T25 cm2 flask) | 27 ± 1 without CO2 | Schneider’s insect medium + 10% HI-FBS + 1% Pen/Strep |
| U4.4 | 2.5 × 107 cells/flask (T25 cm2 flask) | 27 ± 1 without CO2 | Leibowitz’s L-15 GlutaMAX™ + 10% HI-FBS + 2% TPB + 1% Pen/Strep |
| RAE25 | 2.4 × 106 cells/tube (5.5 cm2 flat-sided tubes) | 32 ± 1 without CO2 | L-15 GlutaMAX™ + 10% HI-FBS + 5% TPB |
| HAE/CTVM9 | 2.4 × 106 cells/tube(5.5 cm2 flat-sided tubes) | 32 ± 1 without CO2 | L-15/H-Lac + 20% HI-FBS |
| Cell Line/Organism | rBTV-1 | BTV-26 | rBTV-126S2, S6, S7 |
|---|---|---|---|
| BSR (Hamster–fibroblasts) | + | + | + 1 |
| BFA (Bovine–aorta endothelium) | + | + | NA |
| KC (C. sonorensis midge–larvae) | + | - | - |
| U4.4 (A. albopictus mosquito–) | + | - | NA |
| RAE25 (R. appendiculatus tick–) | - | - | NA |
| HAE/CTVM9 (H. anatolicum tick–) | - | - | NA |
| Adult C. sonorensis midges | + | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guimerà Busquets, M.; Pullinger, G.D.; Darpel, K.E.; Cooke, L.; Armstrong, S.; Simpson, J.; Palmarini, M.; Fragkoudis, R.; Mertens, P.P.C. An Early Block in the Replication of the Atypical Bluetongue Virus Serotype 26 in Culicoides Cells Is Determined by Its Capsid Proteins. Viruses 2021, 13, 919. https://doi.org/10.3390/v13050919
Guimerà Busquets M, Pullinger GD, Darpel KE, Cooke L, Armstrong S, Simpson J, Palmarini M, Fragkoudis R, Mertens PPC. An Early Block in the Replication of the Atypical Bluetongue Virus Serotype 26 in Culicoides Cells Is Determined by Its Capsid Proteins. Viruses. 2021; 13(5):919. https://doi.org/10.3390/v13050919
Chicago/Turabian StyleGuimerà Busquets, Marc, Gillian D. Pullinger, Karin E. Darpel, Lyndsay Cooke, Stuart Armstrong, Jennifer Simpson, Massimo Palmarini, Rennos Fragkoudis, and Peter P. C. Mertens. 2021. "An Early Block in the Replication of the Atypical Bluetongue Virus Serotype 26 in Culicoides Cells Is Determined by Its Capsid Proteins" Viruses 13, no. 5: 919. https://doi.org/10.3390/v13050919
APA StyleGuimerà Busquets, M., Pullinger, G. D., Darpel, K. E., Cooke, L., Armstrong, S., Simpson, J., Palmarini, M., Fragkoudis, R., & Mertens, P. P. C. (2021). An Early Block in the Replication of the Atypical Bluetongue Virus Serotype 26 in Culicoides Cells Is Determined by Its Capsid Proteins. Viruses, 13(5), 919. https://doi.org/10.3390/v13050919