
Molecular Epidemiology and Whole-Genome Analysis of Bovine Foamy Virus in Japan


Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples
2.2. DNA Extraction and Nested PCR
2.3. Whole-Genome Amplification, Library Preparation and Next-Generation Sequencing
2.4. Data Analysis of Next-Generation Sequencing
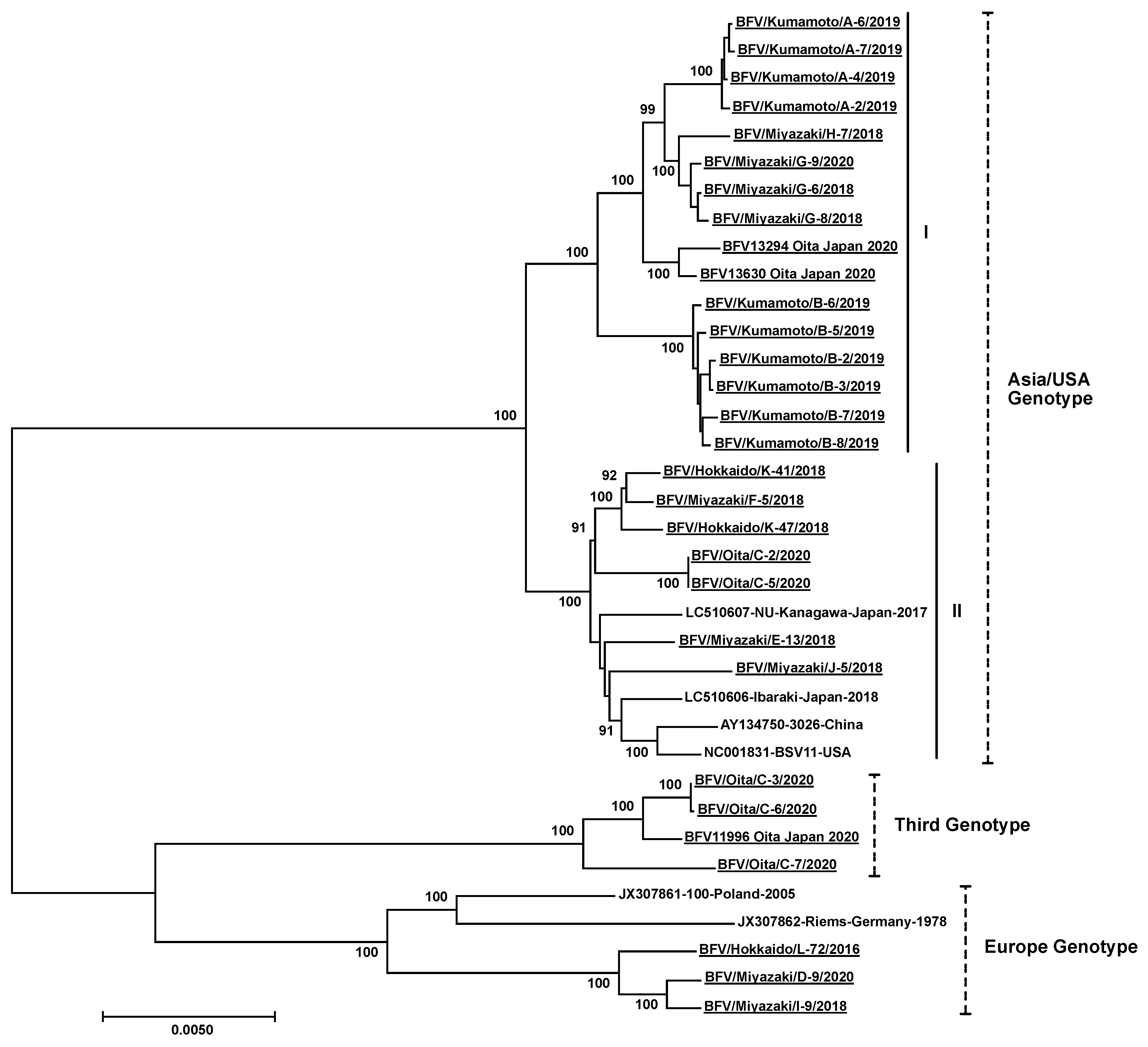
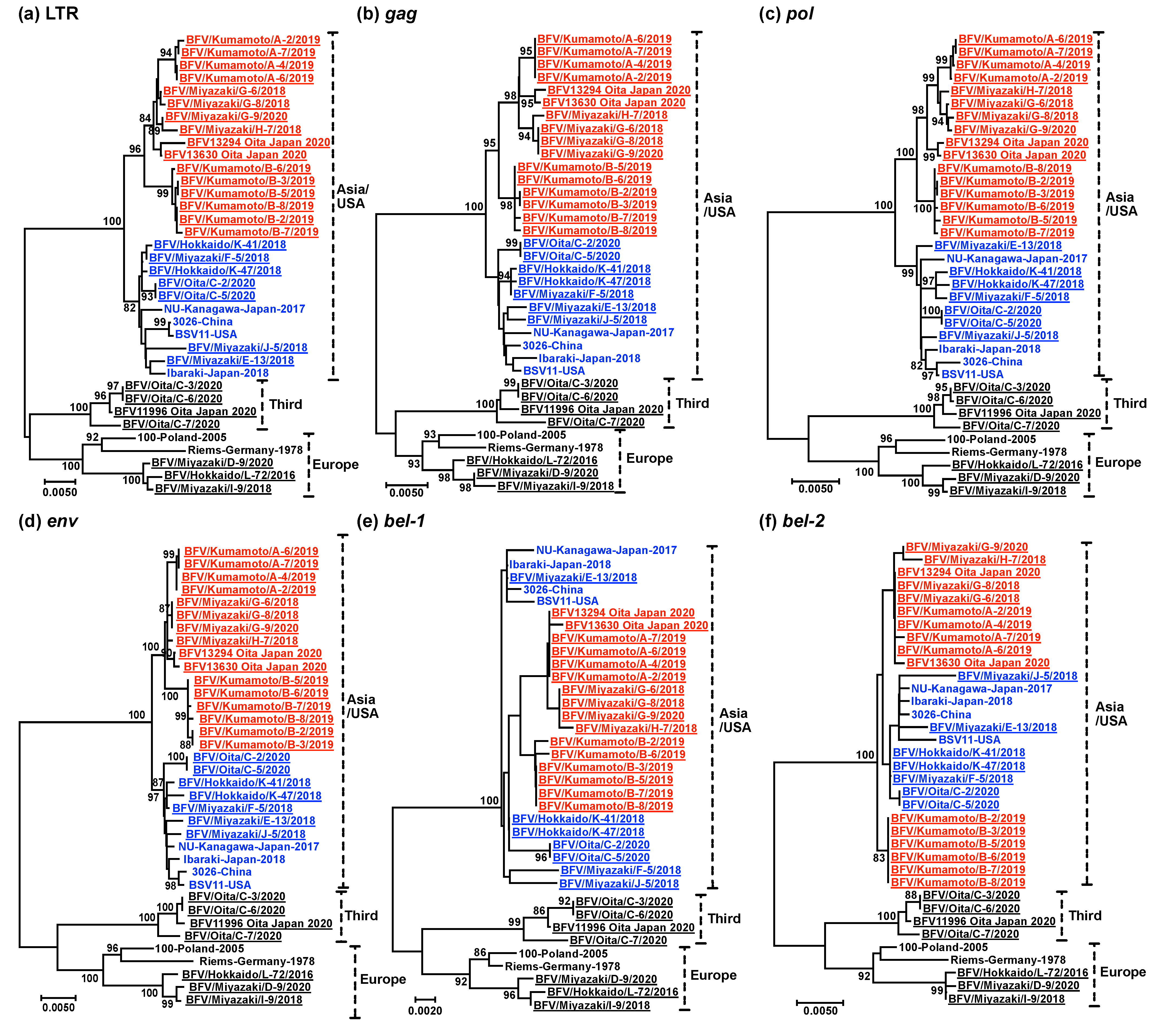
2.5. Phylogenetic Analysis
2.6. Statistical Analysis
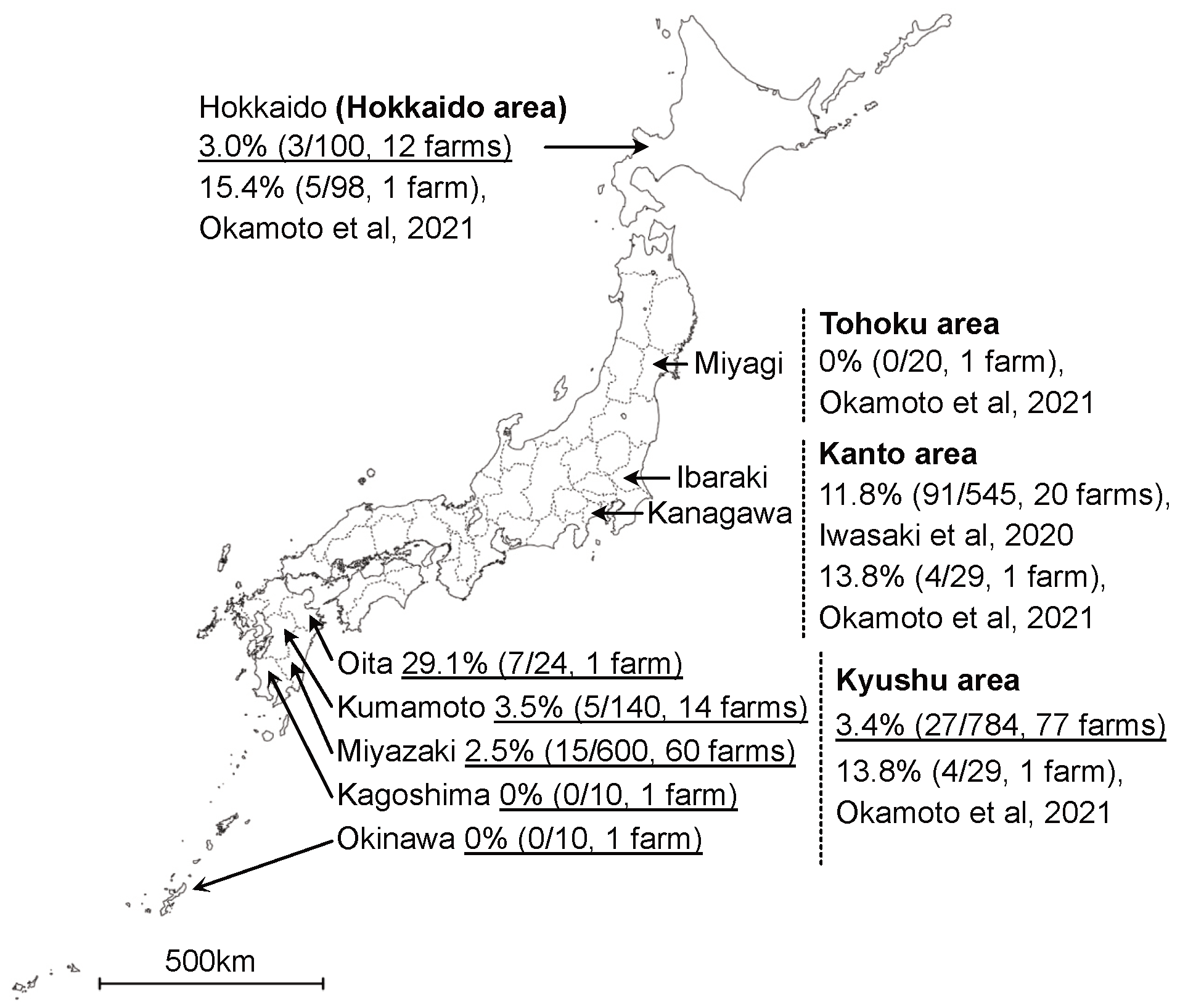
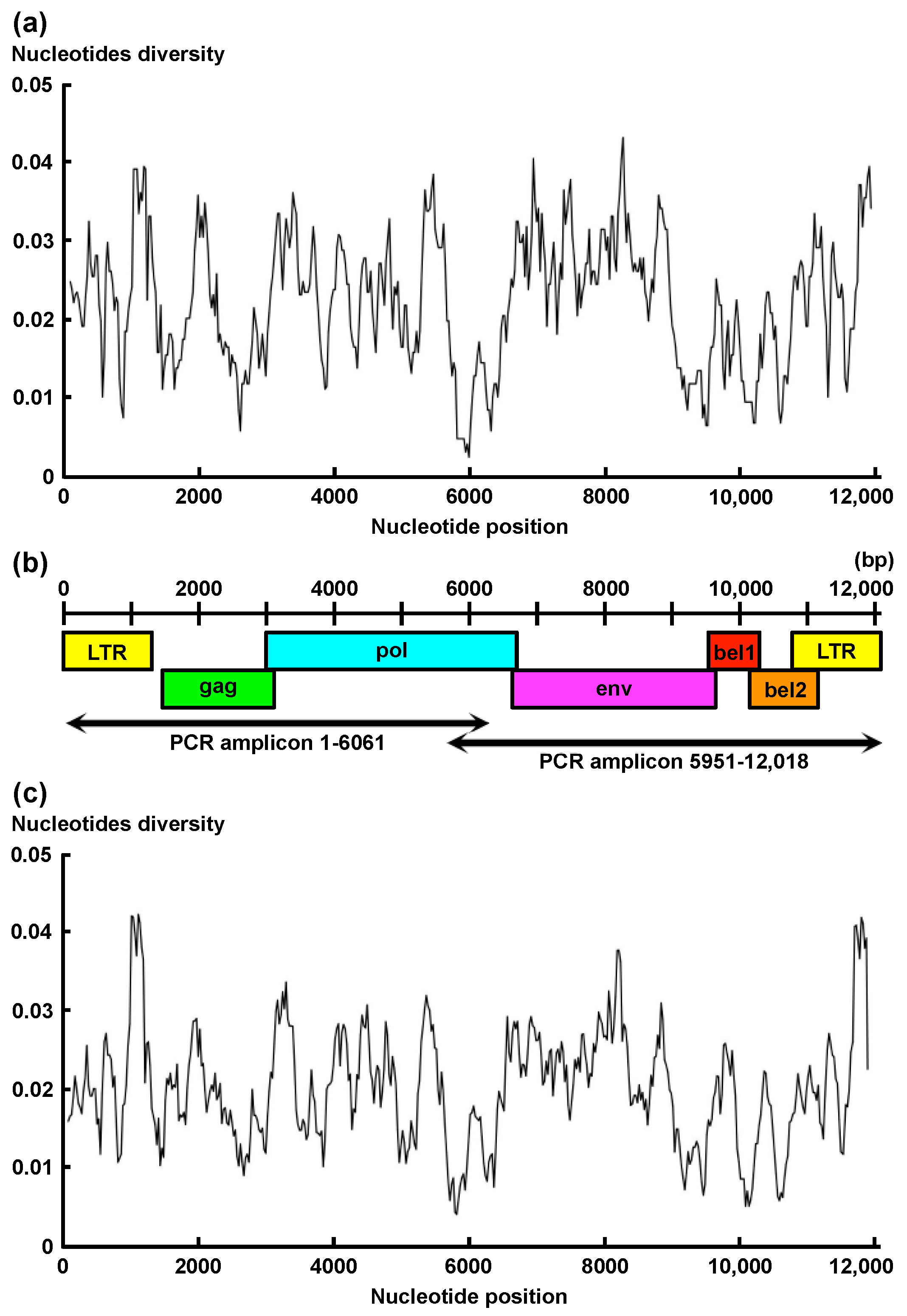
3. Results
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Khan, A.S.; Bodem, J.; Buseyne, F.; Gessain, A.; Johnson, W.; Kuhn, J.H.; Kuzmak, J.; Lindemann, D.; Linial, M.L.; Löchelt, M.; et al. Spumaretroviruses: Updated taxonomy and nomenclature. Virology 2018, 516, 158–164. [Google Scholar] [CrossRef]
- Tan, J.; Qiao, W.; Xu, F.; Han, H.; Chen, Q.; Geng, Y. Dimerization of BTas is required for the transactivational activity of bovine foamy virus. Virology 2008, 376, 236–241. [Google Scholar] [CrossRef] [Green Version]
- Saïb, A.; de Thé, H. Molecular biology of the human foamy virus. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1996, 13, S254–S260. [Google Scholar] [CrossRef]
- Materniak-Kornas, M.; Tan, J.; Heit-Mondrzyk, A.; Hotz-Wagenblatt, A.; Löchelt, M. Bovine foamy virus: Shared and unique molecular features in vitro and in vivo. Viruses 2019, 11, 1084. [Google Scholar] [CrossRef] [Green Version]
- Löchelt, M.; Romen, F.; Bastone, P.; Muckenfuss, H.; Kirchner, N.; Kim, Y.B.; Truyen, U.; Rösler, U.; Battenberg, M.; Saib, A.; et al. The antiretroviral activity of APOBEC3 is inhibited by the foamy virus accessory Bet protein. Proc. Natl. Acad. Sci. USA 2005, 102, 7982–7987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.A.; Wiegand, H.L.; Moore, M.D.; Schäfer, A.; McClure, M.O.; Cullen, B.R. Foamy virus Bet proteins function as novel inhibitors of the APOBEC3 family of innate antiretroviral defense factors. J. Virol. 2005, 79, 8724–8731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, W.; Stricker, E.; Hotz-Wagenblatt, A.; Heit-Mondrzyk, A.; Pougialis, G.; Hugo, A.; Kuźmak, J.; Materniak-Kornas, M.; Löchelt, M. Functional analyses of bovine foamy virus-encoded miRNAs reveal the importance of a defined miRNA for virus replication and host-virus interaction. Viruses 2020, 12, 1250. [Google Scholar] [CrossRef] [PubMed]
- Whisnant, A.W.; Kehl, T.; Bao, Q.; Materniak, M.; Kuzmak, J.; Löchelt, M.; Cullen, B.R. Identification of novel, highly expressed retroviral microRNAs in cells infected by bovine foamy virus. J. Virol. 2014, 88, 4679–4686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falcone, V.; Schweizer, M.; Neumann-Haefelin, D. Replication of primate foamy viruses in natural and experimental hosts. Curr. Top. Microbiol. Immunol. 2003, 277, 161–180. [Google Scholar] [CrossRef]
- Meiering, C.D.; Linial, M.L. Historical perspective of foamy virus epidemiology and infection. Clin. Microbiol. Rev. 2001, 14, 165–176. [Google Scholar] [CrossRef] [Green Version]
- Alke, A.; Schwantes, A.; Zemba, M.; Flügel, R.M.; Löchelt, M. Characterization of the humoral immune response and virus replication in cats experimentally infected with feline foamy virus. Virology 2000, 275, 170–176. [Google Scholar] [CrossRef] [PubMed]
- Falcone, V.; Leupold, J.; Clotten, J.; Urbanyi, E.; Herchenröder, O.; Spatz, W.; Volk, B.; Böhm, N.; Toniolo, A.; Neumann-Haefelin, D.; et al. Sites of simian foamy virus persistence in naturally infected African green monkeys: Latent provirus is ubiquitous, whereas viral replication is restricted to the oral mucosa. Virology 1999, 257, 7–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, R.H.; de la Rosa, J.; Abher, I.; Kertayadnya, I.G.; Entwistle, K.W.; Fordyce, G.; Holroyd, R.G. Epidemiological studies of bovine spumavirus. Vet. Microbiol. 1988, 16, 25–33. [Google Scholar] [CrossRef]
- Romen, F.; Backes, P.; Materniak, M.; Sting, R.; Vahlenkamp, T.W.; Riebe, R.; Pawlita, M.; Kuzmak, J.; Löchelt, M. Serological detection systems for identification of cows shedding bovine foamy virus via milk. Virology 2007, 364, 123–131. [Google Scholar] [CrossRef] [Green Version]
- Materniak-Kornas, M.; Osiński, Z.; Rudzki, M.; Kuźmak, J. Development of a recombinant protein-based ELISA for detection of antibodies against bovine foamy virus. J. Vet. Res. 2017, 61, 247–252. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, R.M.; Pollari, F.L.; McNab, W.B.; Jefferson, B. A serological survey of bovine syncytial virus in Ontario: Associations with bovine leukemia and immunodeficiency-like viruses, production records, and management practices. Can. J. Vet. Res. 1995, 59, 271–278. [Google Scholar] [PubMed]
- Hachiya, Y.; Kimura, K.; Oguma, K.; Ono, M.; Horikita, T.; Sentsui, H. Isolation of bovine foamy virus in Japan. J. Vet. Med. Sci. 2018, 80, 1604–1609. [Google Scholar] [CrossRef]
- Iwasaki, R.; Nakagiri, Y.; Yaguchi, Y.; Oguma, K.; Ono, M.; Horikita, T.; Sentsui, H. Survey of bovine foamy virus infection among cattle in Japan and comparison with bovine leukemia virus infection. J. Vet. Med. Sci. 2020, 82, 615–618. [Google Scholar] [CrossRef]
- Okamoto, M.; Oguma, K.; Yamashita-Kawanishi, N.; Ichijo, T.; Hatama, S.; Endo, M.; Ishikawa, M.; Haga, T. Genomic characterization and distribution of bovine foamy virus in Japan. J. Vet. Med. Sci. 2020, 82, 1607–1613. [Google Scholar] [CrossRef] [PubMed]
- Switzer, W.M.; Salemi, M.; Shanmugam, V.; Gao, F.; Cong, M.E.; Kuiken, C.; Bhullar, V.; Beer, B.E.; Vallet, D.; Gautier-Hion, A.; et al. Ancient co-speciation of simian foamy viruses and primates. Nature 2005, 434, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Katzourakis, A.; Gifford, R.J.; Tristem, M.; Gilbert, M.T.P.; Pybus, O.G. Macroevolution of complex retroviruses. Science 2009, 325, 1512. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.A.; Arumugam, P.; Pillis, D.M.; Loberg, A.; Nasimuzzaman, M.; Lynn, D.; van der Loo, J.C.M.; Dexheimer, P.J.; Keddache, M.; Bauer, T.R., Jr.; et al. Foamy virus vector carries a strong insulator in its long terminal repeat which reduces its genotoxic potential. J. Virol. 2018, 92, e01639-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sweeney, N.P.; Meng, J.; Patterson, H.; Morgan, J.E.; McClure, M. Delivery of large transgene cassettes by foamy virus vector. Sci. Rep. 2017, 7, 8085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ledesma-Feliciano, C.; Hagen, S.; Troyer, R.; Zheng, X.; Musselman, E.; Lukic, D.S.; Franke, A.M.; Maeda, D.; Zielonka, J.; Münk, C.; et al. Replacement of feline foamy virus bet by feline immunodeficiency virus vif yields replicative virus with novel vaccine candidate potential. Retrovirology 2018, 15, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto-Gotoh, A.; Kitao, K.; Miyazawa, T. Persistent infection of simian foamy virus derived from the Japanese macaque leads to the high-level expression of microRNA that resembles the miR-1 microRNA precursor family. Microbes Environ. 2020, 35, ME19130. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Sato, Y.; Mekata, H.; Sudaryatma, P.E.; Kirino, Y.; Yamamoto, S.; Ando, S.; Sugimoto, T.; Okabayashi, T. Isolation of severe fever with thrombocytopenia syndrome virus from various tick species in area with human severe fever with thrombocytopenia syndrome cases. Vector Borne Zoonotic Dis. 2021, 5, 378–384. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Ruckerbauer, G.M.; Sugden, E.A.; Bouillant, A.M. A comparison of the bovine leukemia and bovine syncytial virus status in utero-tubal cells recovered from fluids used to flush the uterus and oviducts of BLV-infected, superovulated cattle. Ann. Rech. Vet. 1988, 19, 19–26. [Google Scholar]
- Schweizer, M.; Falcone, V.; Gänge, J.; Turek, R.; Neumann-Haefelin, D. Simian foamy virus isolated from an accidentally infected human individual. J. Virol. 1997, 71, 4821–4824. [Google Scholar] [CrossRef] [Green Version]
- Mekata, H.; Minamino, T.; Mikurino, Y.; Yamamoto, M.; Yoshida, A.; Nonaka, N.; Horii, Y. Evaluation of the natural vertical transmission of Theileria orientalis. Vet. Parasitol. 2018, 263, 1–4. [Google Scholar] [CrossRef]
- Hechler, T.; Materniak, M.; Kehl, T.; Kuzmak, J.; Löchelt, M. Complete genome sequences of two novel European clade bovine foamy viruses from Germany and Poland. J. Virol. 2012, 86, 10905–10906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, P.B. Taxonomic features of seven serotypes of simian and ape foamy viruses. Infect. Immun. 1971, 3, 793–799. [Google Scholar] [CrossRef] [Green Version]
- Winkler, I.G.; Flügel, R.M.; Löchelt, M.; Flower, R.L.P. Detection and molecular characterization of feline foamy virus serotypes in naturally infected cats. Virology 1998, 247, 144–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mekata, H.; Hamabe, S.; Sudaryatma, P.E.; Kobayashi, I.; Kanno, T.; Okabayashi, T. Molecular epidemiological survey and phylogenetic analysis of bovine respiratory coronavirus in Japan from 2016 to 2018. J. Vet. Med. Sci. 2020, 82, 726–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bao, Q.; Hipp, M.; Hugo, A.; Lei, J.; Liu, Y.; Kehl, T.; Hechler, T.; Löchelt, M. In vitro evolution of bovine foamy virus variants with enhanced cell-free virus titers and transmission. Viruses 2015, 7, 5855–5874. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.; Hotz-Wagenblatt, A.; Betts, M.J.; Hipp, M.; Hugo, A.; Pougialis, G.; Lei-Rossmann, J.; Löchelt, M. Shared and cell type-specific adaptation strategies of Gag and Env yield high titer bovine foamy virus variants. Infect. Genetics. Evol. 2020, 82, 104287. [Google Scholar] [CrossRef]
- Bing, T.; Wu, K.; Cui, X.; Shao, P.; Zhang, Q.; Bai, X.; Tan, J.; Qiao, W. Identification and functional characterization of Bet protein as a negative regulator of BFV3026 replication. Virus Genes 2014, 48, 464–473. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Prefecture | Area | No. of Positive Farms/Tests (Positive Rate) | No. of Positive Cattle/Tests (Positive Rate) |
|---|---|---|---|
| Miyazaki | Kyushu | 8/60 (13.3%) | 15/600 (2.5%) |
| Kumamoto | Kyushu | 1/14 (7.1%) | 5/140 (3.5%) |
| Oita | Kyushu | 1/1 (100%) | 7/24 (29.1%) |
| Okinawa | Kyushu | 0/1 (0%) | 0/10 (0%) |
| Kagoshima | Kyushu | 0/1 (0%) | 0/10 (0%) |
| Hokkaido | Hokkaido | 2/12 (16.6%) | 3/100 (3.0%) |
| Total | 12/89 (13.4%) | 30/884 (3.4%) |
| Age (Months) | No. of Samples | No. of Positive Cattle (%) |
|---|---|---|
| 0–48 | 255 | 0 (0) |
| 48–84 | 265 | 13 (4.9) |
| 84–120 | 221 | 9 (4.3) |
| >120 | 110 | 7 (6.7) |
| Unknown | 33 | 1 (3.0) |
| Total | 884 | 30 (3.4) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mekata, H.; Okagawa, T.; Konnai, S.; Miyazawa, T. Molecular Epidemiology and Whole-Genome Analysis of Bovine Foamy Virus in Japan. Viruses 2021, 13, 1017. https://doi.org/10.3390/v13061017
Mekata H, Okagawa T, Konnai S, Miyazawa T. Molecular Epidemiology and Whole-Genome Analysis of Bovine Foamy Virus in Japan. Viruses. 2021; 13(6):1017. https://doi.org/10.3390/v13061017
Chicago/Turabian StyleMekata, Hirohisa, Tomohiro Okagawa, Satoru Konnai, and Takayuki Miyazawa. 2021. "Molecular Epidemiology and Whole-Genome Analysis of Bovine Foamy Virus in Japan" Viruses 13, no. 6: 1017. https://doi.org/10.3390/v13061017
APA StyleMekata, H., Okagawa, T., Konnai, S., & Miyazawa, T. (2021). Molecular Epidemiology and Whole-Genome Analysis of Bovine Foamy Virus in Japan. Viruses, 13(6), 1017. https://doi.org/10.3390/v13061017