
Correlating qRT-PCR, dPCR and Viral Titration for the Identification and Quantification of SARS-CoV-2: A New Approach for Infection Management

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
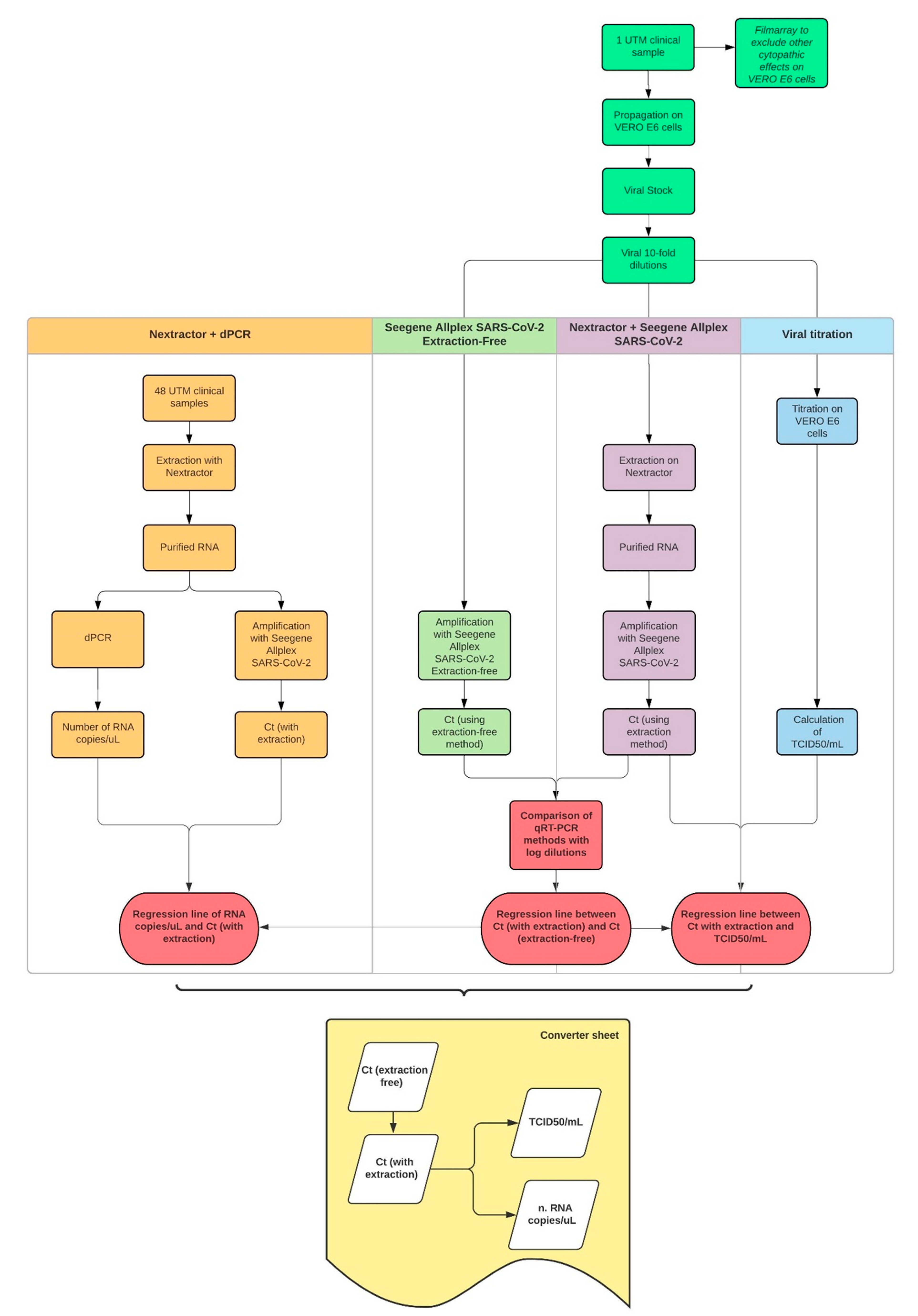
2. Materials and Methods
2.1. Cell Line and Virus
2.2. Viral Titration
2.3. Nucleic Acid Quantification
2.3.1. Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
2.3.2. Digital Polymerase Chain Reaction (dPCR)
3. Results
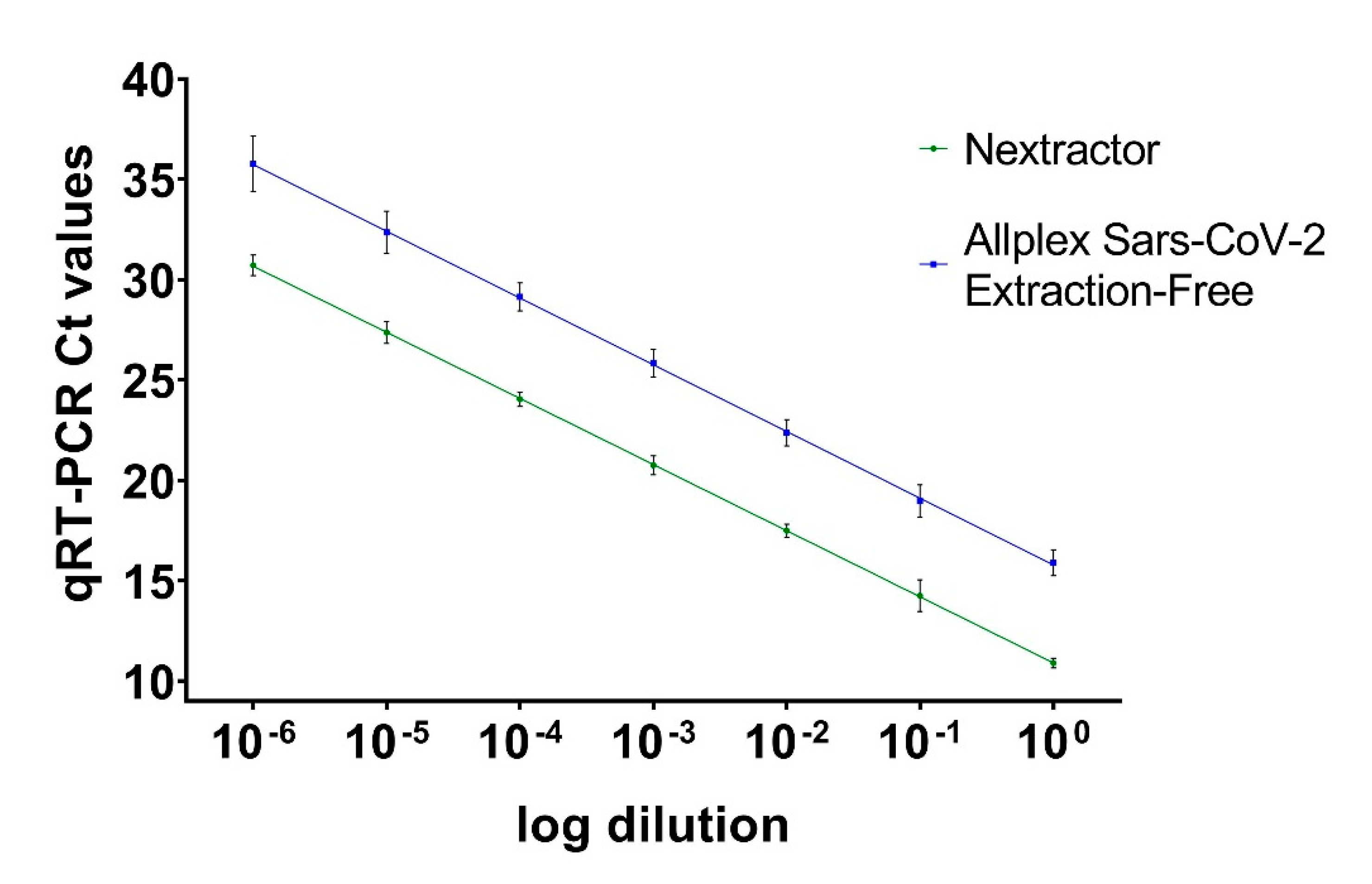
3.1. Comparison of qRT-PCR Methods
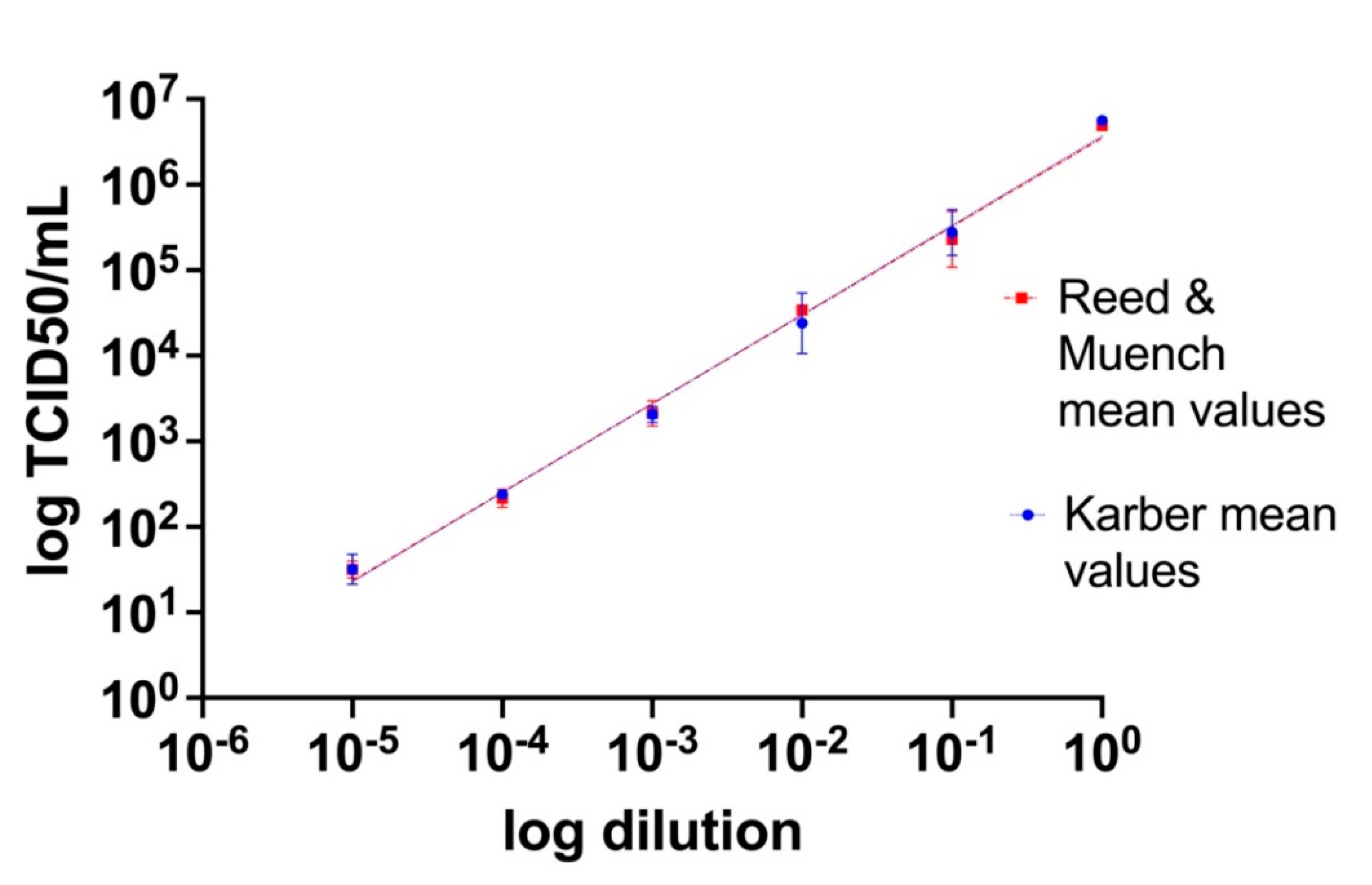
3.2. Calculation of Tissue Culture Infecting Dose per mL (TCID50/mL)
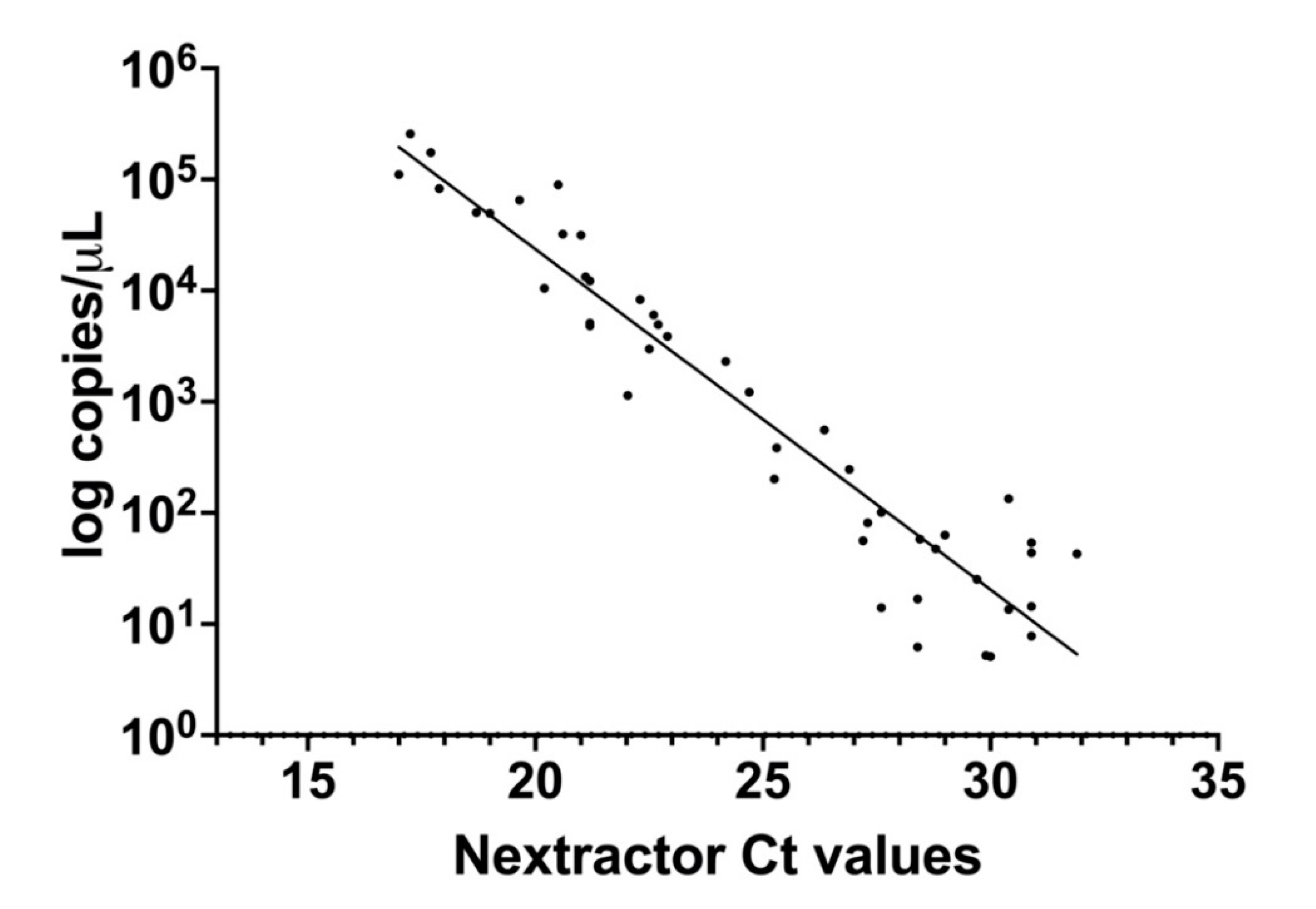
3.3. Correlation between dPCR and qRT-PCR
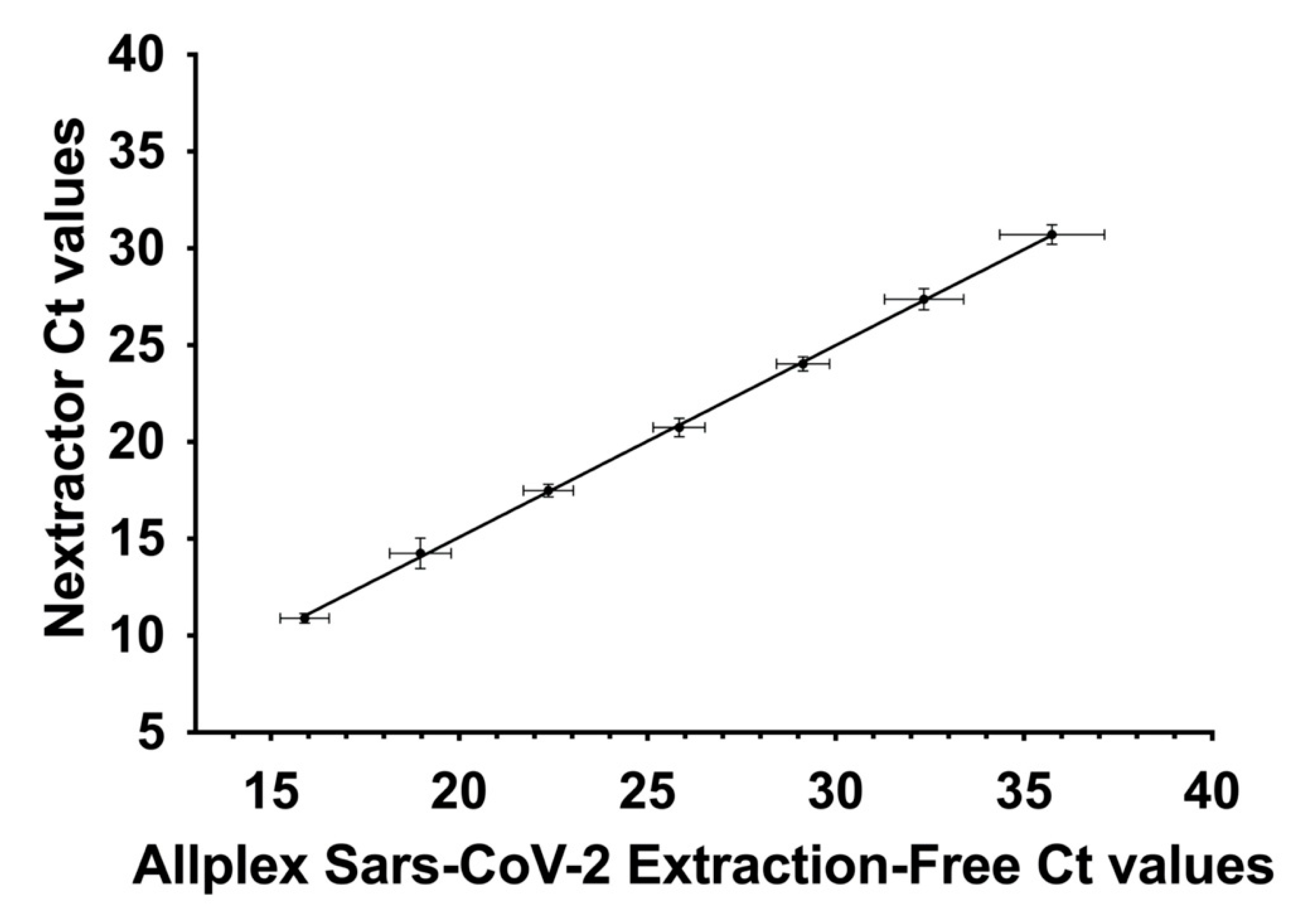
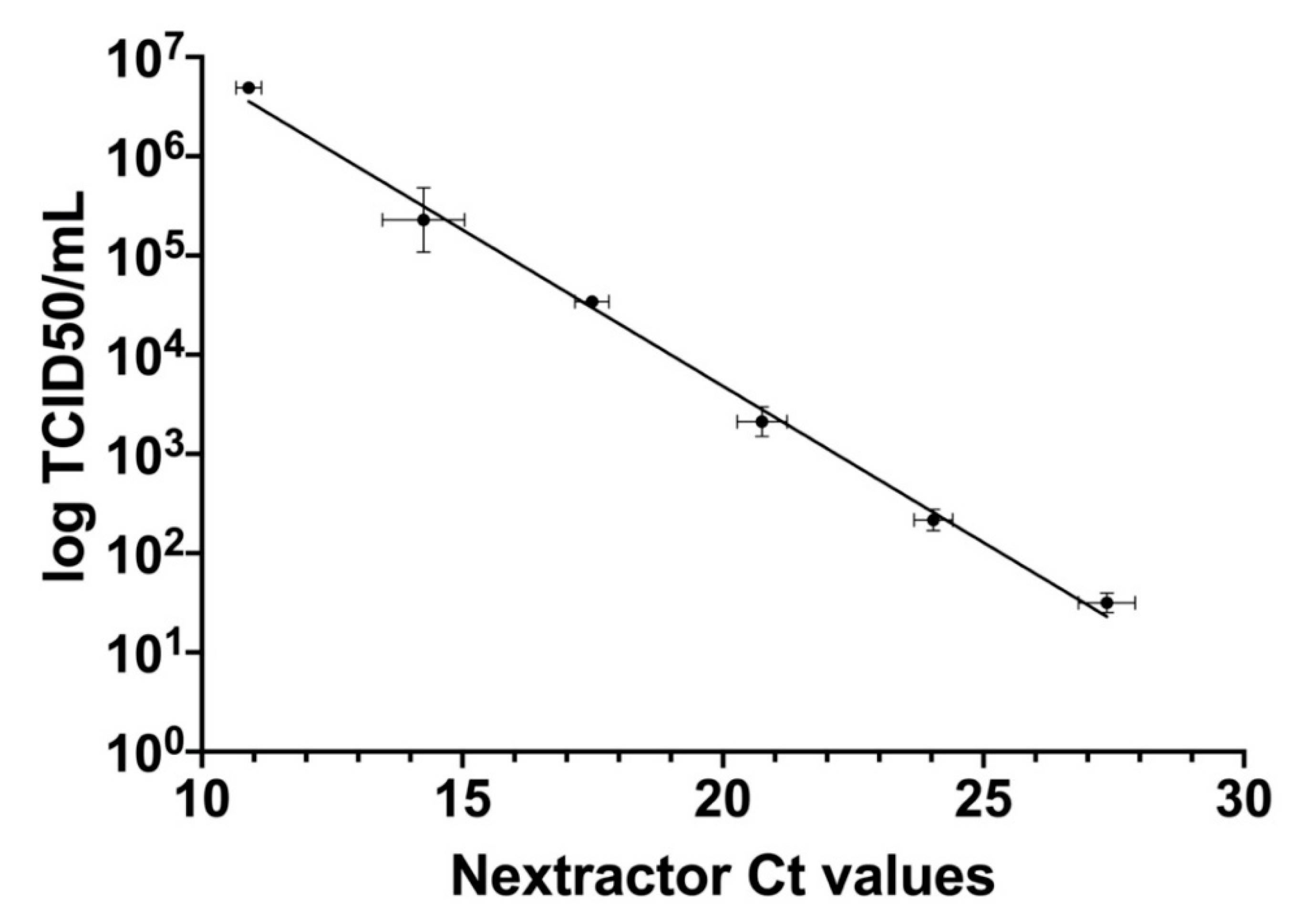
3.4. Correlation between qRT-PCR and TCID50/mL
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent insights into emerging coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A Novel Coronavirus Associated with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Wang, L.-F.; Shi, Z.; Zhang, S.; Field, H.; Daszak, P.; Eaton, B.T. Review of Bats and SARS. Emerg. Infect. Dis. 2006, 12, 1834–1840. [Google Scholar] [CrossRef]
- Zaki, A.; Van Boheemen, S.; Bestebroer, T.; Osterhaus, A.; Fouchier, R. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Ithete, N.; Stoffberg, S.; Corman, V.M.; Cottontail, V.M.; Richards, L.R.; Schoeman, C.; Drosten, C.; Drexler, J.F.; Preiser, W. Close Relative of Human Middle East Respiratory Syndrome Coronavirus in Bat, South Africa. Emerg. Infect. Dis. 2013, 19, 1697–1699. [Google Scholar] [CrossRef]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.J.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Butt, K.M.; et al. Isolation and Characterization of Viruses Related to the SARS Coronavirus from Animals in Southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wong, S.-K.; Li, F.; Kuhn, J.H.; Huang, I.-C.; Choe, H.; Farzan, M. Animal Origins of the Severe Acute Respiratory Syndrome Coronavirus: Insight from ACE2-S-Protein Interactions. J. Virol. 2006, 80, 4211–4219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolles, M.; Donaldson, E.; Baric, R. SARS-CoV and emergent coronaviruses: Viral determinants of interspecies transmission. Curr. Opin. Virol. 2011, 1, 624–634. [Google Scholar] [CrossRef]
- Chan, J.F.; To, K.K.; Tse, H.; Jin, D.Y.; Yuen, K.Y. Interspecies transmission and emergence of novel viruses: Lessons from bats and birds. Trends Microbiol. 2013, 21, 544–555. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Lian, X.; Su, X.; Wu, W.; Marraro, G.A.; Zeng, Y. From SARS and MERS to COVID-19: A brief summary and comparison of severe acute respiratory infections caused by three highly pathogenic human coronaviruses. Respir. Res. 2020, 21, 1–14. [Google Scholar] [CrossRef]
- Rothan, H.A.; Byrareddy, S.N. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J. Autoimmun. 2020, 109, 102433. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. 2020. Available online: https://covid19.who.int/ (accessed on 28 January 2021).
- Stelzer-Braid, S.; Walker, G.J.; Aggarwal, A.; Isaacs, S.R.; Yeang, M.; Naing, Z.; Stella, A.O.; Turville, S.G.; Rawlinson, W.D. Virus isolation of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) for diagnostic and research purposes. Pathology 2020, 52, 760–763. [Google Scholar] [CrossRef] [PubMed]
- Lei, C.; Yang, J.; Hu, J.; Sun, X. On the Calculation of TCID50 for Quantitation of Virus Infectivity. Virol. Sin. 2021, 36, 141–144. [Google Scholar] [CrossRef]
- Amanat, F.; White, K.M.; Miorin, L.; Strohmeier, S.; McMahon, M.; Meade, P.; Liu, W.; Albrecht, R.A.; Simon, V.; Martinez-Sobrido, L.; et al. An In Vitro Microneutralization Assay for SARS-CoV-2 Serology and Drug Screening. Curr. Protoc. Microbiol. 2020, 58, e108. [Google Scholar] [CrossRef] [PubMed]
- Kuypers, J.; Jerome, K.R. Applications of Digital PCR for Clinical Microbiology. J. Clin. Microbiol. 2017, 55, 1621–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salipante, S.J.; Jerome, K.R. Digital PCR—An Emerging Technology with Broad Applications in Microbiology. Clin. Chem. 2019, 66, 117–123. [Google Scholar] [CrossRef]
- Ogando, N.S.; Dalebout, T.J.; Zevenhoven-Dobbe, J.C.; Limpens, R.W.; Van Der Meer, Y.; Caly, L.; Druce, J.; De Vries, J.J.C.; Kikkert, M.; Bárcena, M.; et al. SARS-coronavirus-2 replication in Vero E6 cells: Replication kinetics, rapid adaptation and cytopathology. J. Gen. Virol. 2020, 101, 925–940. [Google Scholar] [CrossRef]
- Leist, S.R.; Schäfer, A.; Martinez, D.R. Cell and animal models of SARS-CoV-2 pathogenesis and immunity. Dis. Model. Mech. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K. In Vitro and Animal Models for SARS-CoV-2 research. Trends Pharmacol. Sci. 2020, 41, 513–517. [Google Scholar] [CrossRef]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Smith, M.R.; Schirtzinger, E.E.; Wilson, W.C.; Davis, A.S. Rift Valley Fever Virus: Propagation, Quantification, and Storage. Curr. Protoc. Microbiol. 2019, 55, e92. [Google Scholar] [CrossRef]
- GraphPad Prism 9 User Guide. Available online: www.graphpad.com (accessed on 2 January 2021).
- Alexandersen, S.; Chamings, A.; Bhatta, T.R. SARS-CoV-2 genomic and subgenomic RNAs in diagnostic samples are not an indicator of active replication. Nat. Commun. 2020, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Burnett, L.; McQueen, M.J.; Jonsson, J.J.; Torricelli, F. IFCC Taskforce on Ethics. IFCC Position Paper: Report of the IFCC Taskforce on Ethics: Introduction and framework. Clin. Chem. Lab. Med. 2007, 45, 1098–1104. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandolini, M.; Taddei, F.; Marino, M.M.; Grumiro, L.; Scalcione, A.; Turba, M.E.; Gentilini, F.; Fantini, M.; Zannoli, S.; Dirani, G.; et al. Correlating qRT-PCR, dPCR and Viral Titration for the Identification and Quantification of SARS-CoV-2: A New Approach for Infection Management. Viruses 2021, 13, 1022. https://doi.org/10.3390/v13061022
Brandolini M, Taddei F, Marino MM, Grumiro L, Scalcione A, Turba ME, Gentilini F, Fantini M, Zannoli S, Dirani G, et al. Correlating qRT-PCR, dPCR and Viral Titration for the Identification and Quantification of SARS-CoV-2: A New Approach for Infection Management. Viruses. 2021; 13(6):1022. https://doi.org/10.3390/v13061022
Chicago/Turabian StyleBrandolini, Martina, Francesca Taddei, Maria Michela Marino, Laura Grumiro, Agata Scalcione, Maria Elena Turba, Fabio Gentilini, Michela Fantini, Silvia Zannoli, Giorgio Dirani, and et al. 2021. "Correlating qRT-PCR, dPCR and Viral Titration for the Identification and Quantification of SARS-CoV-2: A New Approach for Infection Management" Viruses 13, no. 6: 1022. https://doi.org/10.3390/v13061022
APA StyleBrandolini, M., Taddei, F., Marino, M. M., Grumiro, L., Scalcione, A., Turba, M. E., Gentilini, F., Fantini, M., Zannoli, S., Dirani, G., & Sambri, V. (2021). Correlating qRT-PCR, dPCR and Viral Titration for the Identification and Quantification of SARS-CoV-2: A New Approach for Infection Management. Viruses, 13(6), 1022. https://doi.org/10.3390/v13061022