
Requirement of Fc-Fc Gamma Receptor Interaction for Antibody-Based Protection against Emerging Virus Infections


Abstract
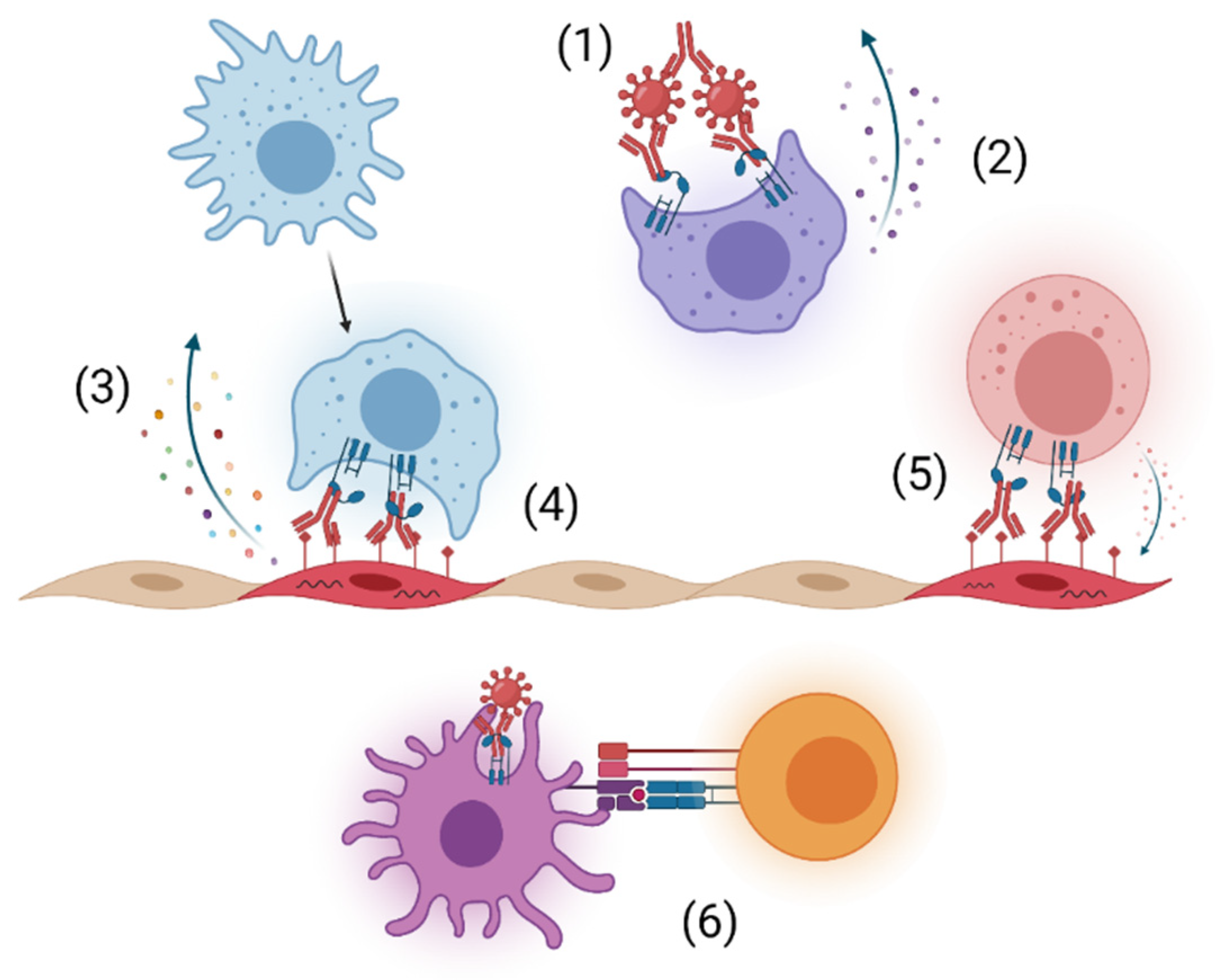
:1. Introduction
2. Approaches to Study Fc Effector Functions In Vivo
2.1. IgG Subclass and FcγRs
2.2. Fc Mutations
2.3. Knockout and Transgenic Mice
3. Optimal mAb Efficacy In Vivo through Fc-Effector Functions
3.1. Influenza Virus
3.2. Coronaviruses
3.3. Alphaviruses
3.4. Flaviviruses
3.5. Filoviruses
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- WHO Ebola Response Team; Aylward, B.; Barboza, P.; Bawo, L.; Bertherat, E.; Bilivogui, P.; Blake, I.; Brennan, R.; Briand, S.; Chakauya, J.M.; et al. Ebola virus disease in West Africa--the first 9 months of the epidemic and forward projections. N. Engl. J. Med. 2014, 371, 1481–1495. [Google Scholar]
- Paules, C.I.; Marston, H.D.; Fauci, A.S. Coronavirus Infections-More Than Just the Common Cold. JAMA 2020, 323, 707–708. [Google Scholar] [CrossRef] [Green Version]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef] [Green Version]
- Gatherer, D.; Kohl, A. Zika virus: A previously slow pandemic spreads rapidly through the Americas. J. Gen. Virol. 2016, 97, 269–273. [Google Scholar] [CrossRef] [Green Version]
- Cutler, D.M.; Summers, L.H. The COVID-19 Pandemic and the $16 Trillion Virus. JAMA 2020, 324, 1495–1496. [Google Scholar] [CrossRef]
- Fonseca, M.H.G.; Furtado, G.P.; Bezerra, M.R.L.; Pontes, L.Q.; Fernandes, C.F.C. Boosting half-life and effector functions of therapeutic antibodies by Fc-engineering: An interaction-function review. Int. J. Biol. Macromol. 2018, 119, 306–311. [Google Scholar] [CrossRef]
- Irani, V.; Guy, A.J.; Andrew, D.; Beeson, J.G.; Ramsland, P.A.; Richards, J.S. Molecular properties of human IgG subclasses and their implications for designing therapeutic monoclonal antibodies against infectious diseases. Mol. Immunol. 2015, 67, 171–182. [Google Scholar] [CrossRef]
- Balsitis, S.J.; Williams, K.L.; Lachica, R.; Flores, D.; Kyle, J.L.; Mehlhop, E.; Johnson, S.; Diamond, M.S.; Beatty, P.R.; Harris, E. Lethal antibody enhancement of dengue disease in mice is prevented by Fc modification. PLoS Pathog. 2010, 6, e1000790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halstead, S.B.; Mahalingam, S.; Marovich, M.A.; Ubol, S.; Mosser, D.M. Intrinsic antibody-dependent enhancement of microbial infection in macrophages: Disease regulation by immune complexes. Lancet Infect. Dis. 2010, 10, 712–722. [Google Scholar] [CrossRef] [Green Version]
- Huber, V.C.; Lynch, J.M.; Bucher, D.J.; Le, J.; Metzger, D.W. Fc receptor-mediated phagocytosis makes a significant contribution to clearance of influenza virus infections. J. Immunol. 2001, 166, 7381–7388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winkler, E.S.; Gilchuk, P.; Yu, J.; Bailey, A.L.; Chen, R.E.; Chong, Z.; Zost, S.J.; Jang, H.; Huang, Y.; Allen, J.D.; et al. Human neutralizing antibodies against SARS-CoV-2 require intact Fc effector functions for optimal therapeutic protection. Cell 2021, 184, 1804–1820. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.M.; Roy, V.; Gunn, B.M.; Huang, L.; Edeling, M.A.; Mack, M.; Fremont, D.H.; Doranz, B.J.; Johnson, S.; Alter, G.; et al. Optimal therapeutic activity of monoclonal antibodies against chikungunya virus requires Fc-FcgammaR interaction on monocytes. Sci. Immunol. 2019, 4. [Google Scholar] [CrossRef]
- Bournazos, S.; Klein, F.; Pietzsch, J.; Seaman, M.S.; Nussenzweig, M.C.; Ravetch, J.V. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell 2014, 158, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Bournazos, S.; DiLillo, D.J.; Goff, A.J.; Glass, P.J.; Ravetch, J.V. Differential requirements for FcgammaR engagement by protective antibodies against Ebola virus. Proc. Natl. Acad. Sci. USA 2019, 116, 20054–20062. [Google Scholar] [CrossRef] [Green Version]
- Wessel, A.W.; Kose, N.; Bombardi, R.G.; Roy, V.; Chantima, W.; Mongkolsapaya, J.; Edeling, M.A.; Nelson, C.A.; Bosch, I.; Alter, G.; et al. Antibodies targeting epitopes on the cell-surface form of NS1 protect against Zika virus infection during pregnancy. Nat. Commun. 2020, 11, 5278. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Bruhns, P.; Horiuchi, K.; Ravetch, J.V. FcgammaRIV: A novel FcR with distinct IgG subclass specificity. Immunity 2005, 23, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Bolland, S.; Ravetch, J.V. Inhibitory pathways triggered by ITIM-containing receptors. Adv. Immunol. 1999, 72, 149–177. [Google Scholar] [PubMed]
- Dekkers, G.; Bentlage, A.E.H.; Stegmann, T.C.; Howie, H.L.; Lissenberg-Thunnissen, S.; Zimring, J.; Rispens, T.; Vidarsson, G. Affinity of human IgG subclasses to mouse Fc gamma receptors. MAbs 2017, 9, 767–773. [Google Scholar] [CrossRef]
- Bruhns, P.; Jonsson, F. Mouse and human FcR effector functions. Immunol. Rev. 2015, 268, 25–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nimmerjahn, F.; Ravetch, J.V. Fcgamma receptors as regulators of immune responses. Nat. Rev. Immunol. 2008, 8, 34–47. [Google Scholar] [CrossRef]
- Dijstelbloem, H.M.; van de Winkel, J.G.; Kallenberg, C.G. Inflammation in autoimmunity: Receptors for IgG revisited. Trends Immunol. 2001, 22, 510–516. [Google Scholar] [CrossRef]
- Overdijk, M.B.; Verploegen, S.; Ortiz Buijsse, A.; Vink, T.; Leusen, J.H.; Bleeker, W.K.; Parren, P.W. Crosstalk between human IgG isotypes and murine effector cells. J. Immunol. 2012, 189, 3430–3438. [Google Scholar] [CrossRef]
- Ghetie, V.; Ward, E.S.; Vitetta, E.S. Pharmacokinetics of Antibodies and Immunotoxins in Mice and Humans. In Handbook of Anticancer Pharmacokinetics and Pharmacodynamics; Figg, W.D., McLeod, H.L., Eds.; Humana Press: Totowa, NJ, USA, 2004; pp. 475–498. [Google Scholar]
- Saunders, K.O. Conceptual Approaches to Modulating Antibody Effector Functions and Circulation Half-Life. Front. Immunol. 2019, 10, 1296. [Google Scholar] [CrossRef] [PubMed]
- Arduin, E.; Arora, S.; Bamert, P.R.; Kuiper, T.; Popp, S.; Geisse, S.; Grau, R.; Calzascia, T.; Zenke, G.; Kovarik, J. Highly reduced binding to high and low affinity mouse Fc gamma receptors by L234A/L235A and N297A Fc mutations engineered into mouse IgG2a. Mol. Immunol. 2015, 63, 456–463. [Google Scholar] [CrossRef]
- Walker, M.R.; Lund, J.; Thompson, K.M.; Jefferis, R. Aglycosylation of human IgG1 and IgG3 monoclonal antibodies can eliminate recognition by human cells expressing Fc gamma RI and/or Fc gamma RII receptors. Biochem. J. 1989, 259, 347–353. [Google Scholar] [CrossRef]
- Tao, M.H.; Morrison, S.L. Studies of aglycosylated chimeric mouse-human IgG. Role of carbohydrate in the structure and effector functions mediated by the human IgG constant region. J. Immunol. 1989, 143, 2595–2601. [Google Scholar]
- Latypov, R.F.; Hogan, S.; Lau, H.; Gadgil, H.; Liu, D. Elucidation of acid-induced unfolding and aggregation of human immunoglobulin IgG1 and IgG2 Fc. J. Biol. Chem. 2012, 287, 1381–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horton, H.M.; Bernett, M.J.; Peipp, M.; Pong, E.; Karki, S.; Chu, S.Y.; Richards, J.O.; Chen, H.; Repp, R.; Desjarlais, J.R.; et al. Fc-engineered anti-CD40 antibody enhances multiple effector functions and exhibits potent in vitro and in vivo antitumor activity against hematologic malignancies. Blood 2010, 116, 3004–3012. [Google Scholar] [CrossRef] [Green Version]
- Baudino, L.; Shinohara, Y.; Nimmerjahn, F.; Furukawa, J.; Nakata, M.; Martinez-Soria, E.; Petry, F.; Ravetch, J.V.; Nishimura, S.; Izui, S. Crucial role of aspartic acid at position 265 in the CH2 domain for murine IgG2a and IgG2b Fc-associated effector functions. J. Immunol. 2008, 181, 6664–6669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hezareh, M.; Hessell, A.J.; Jensen, R.C.; van de Winkel, J.G.; Parren, P.W. Effector function activities of a panel of mutants of a broadly neutralizing antibody against human immunodeficiency virus type 1. J. Virol. 2001, 75, 12161–12168. [Google Scholar] [CrossRef] [Green Version]
- Schlothauer, T.; Herter, S.; Koller, C.F.; Grau-Richards, S.; Steinhart, V.; Spick, C.; Kubbies, M.; Klein, C.; Umana, P.; Mossner, E. Novel human IgG1 and IgG4 Fc-engineered antibodies with completely abolished immune effector functions. Protein Eng. Des. Sel. 2016, 29, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Lo, M.; Kim, H.S.; Tong, R.K.; Bainbridge, T.W.; Vernes, J.M.; Zhang, Y.; Lin, Y.L.; Chung, S.; Dennis, M.S.; Zuchero, Y.J.; et al. Effector-attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice. J. Biol. Chem. 2017, 292, 3900–3908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A.A.; Keremane, S.R.; Vielmetter, J.; Bjorkman, P.J. Structural characterization of GASDALIE Fc bound to the activating Fc receptor FcgammaRIIIa. J. Struct. Biol. 2016, 194, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Bournazos, S.; Corti, D.; Virgin, H.W.; Ravetch, J.V. Fc-optimized antibodies elicit CD8 immunity to viral respiratory infection. Nature 2020, 588, 485–490. [Google Scholar] [CrossRef]
- Richards, J.O.; Karki, S.; Lazar, G.A.; Chen, H.; Dang, W.; Desjarlais, J.R. Optimization of antibody binding to FcgammaRIIa enhances macrophage phagocytosis of tumor cells. Mol. Cancer Ther. 2008, 7, 2517–2527. [Google Scholar] [CrossRef] [Green Version]
- Takai, T.; Li, M.; Sylvestre, D.; Clynes, R.; Ravetch, J.V. FcR gamma chain deletion results in pleiotrophic effector cell defects. Cell 1994, 76, 519–529. [Google Scholar] [CrossRef]
- Smith, P.; DiLillo, D.J.; Bournazos, S.; Li, F.; Ravetch, J.V. Mouse model recapitulating human Fcgamma receptor structural and functional diversity. Proc. Natl. Acad. Sci. USA 2012, 109, 6181–6186. [Google Scholar] [CrossRef] [Green Version]
- Takai, T.; Ono, M.; Hikida, M.; Ohmori, H.; Ravetch, J.V. Augmented humoral and anaphylactic responses in Fc gamma RII-deficient mice. Nature 1996, 379, 346–349. [Google Scholar] [CrossRef] [PubMed]
- Hazenbos, W.L.; Gessner, J.E.; Hofhuis, F.M.; Kuipers, H.; Meyer, D.; Heijnen, I.A.; Schmidt, R.E.; Sandor, M.; Capel, P.J.; Daeron, M.; et al. Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity 1996, 5, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Nimmerjahn, F.; Lux, A.; Albert, H.; Woigk, M.; Lehmann, C.; Dudziak, D.; Smith, P.; Ravetch, J.V. FcgammaRIV deletion reveals its central role for IgG2a and IgG2b activity in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 19396–19401. [Google Scholar] [CrossRef] [Green Version]
- Ioan-Facsinay, A.; de Kimpe, S.J.; Hellwig, S.M.; van Lent, P.L.; Hofhuis, F.M.; van Ojik, H.H.; Sedlik, C.; da Silveira, S.A.; Gerber, J.; de Jong, Y.F.; et al. FcgammaRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity 2002, 16, 391–402. [Google Scholar] [CrossRef]
- Kolpe, A.; Schepens, B.; Ye, L.; Staeheli, P.; Saelens, X. Passively transferred M2e-specific monoclonal antibody reduces influenza A virus transmission in mice. Antiviral Res. 2018, 158, 244–254. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Gordan, S.; Lux, A. FcgammaR dependent mechanisms of cytotoxic, agonistic, and neutralizing antibody activities. Trends Immunol. 2015, 36, 325–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- CDC, 2009 H1N1 Pandemic (H1N1pdm09 Virus). Available online: https://www.cdc.gov/flu/pandemic-resources/2009-h1n1-pandemic.html (accessed on 2 April 2021).
- McAuley, J.L.; Gilbertson, B.P.; Trifkovic, S.; Brown, L.E.; McKimm-Breschkin, J.L. Influenza Virus Neuraminidase Structure and Functions. Front. Microbiol. 2019, 10, 39. [Google Scholar] [CrossRef] [Green Version]
- Wu, N.C.; Wilson, I.A. Structural Biology of Influenza Hemagglutinin: An Amaranthine Adventure. Viruses 2020, 12, 1053. [Google Scholar] [CrossRef]
- Kolpe, A.; Schepens, B.; Fiers, W.; Saelens, X. M2-based influenza vaccines: Recent advances and clinical potential. Expert Rev. Vaccines 2017, 16, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Dou, D.; Revol, R.; Ostbye, H.; Wang, H.; Daniels, R. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front. Immunol. 2018, 9, 1581. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Tan, G.S.; Palese, P.; Ravetch, J.V. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat. Med. 2014, 20, 143–151. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Palese, P.; Wilson, P.C.; Ravetch, J.V. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Investig. 2016, 126, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Amanat, F.; Meade, P.; Strohmeier, S.; Krammer, F. Cross-reactive antibodies binding to H4 hemagglutinin protect against a lethal H4N6 influenza virus challenge in the mouse model. Emerg. Microbes Infect. 2019, 8, 155–168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henry Dunand, C.J.; Leon, P.E.; Huang, M.; Choi, A.; Chromikova, V.; Ho, I.Y.; Tan, G.S.; Cruz, J.; Hirsh, A.; Zheng, N.Y.; et al. Both Neutralizing and Non-Neutralizing Human H7N9 Influenza Vaccine-Induced Monoclonal Antibodies Confer Protection. Cell Host Microbe 2016, 19, 800–813. [Google Scholar] [CrossRef] [Green Version]
- Corti, D.; Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011, 333, 850–856. [Google Scholar] [CrossRef]
- Jegaskanda, S.; Job, E.R.; Kramski, M.; Laurie, K.; Isitman, G.; de Rose, R.; Winnall, W.R.; Stratov, I.; Brooks, A.G.; Reading, P.C.; et al. Cross-reactive influenza-specific antibody-dependent cellular cytotoxicity antibodies in the absence of neutralizing antibodies. J. Immunol. 2013, 190, 1837–1848. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Chen, C.J.; Mullarkey, C.E.; Hamilton, J.R.; Wong, C.K.; Leon, P.E.; Uccellini, M.B.; Chromikova, V.; Henry, C.; Hoffman, K.W.; et al. Alveolar macrophages are critical for broadly-reactive antibody-mediated protection against influenza A virus in mice. Nat. Commun. 2017, 8, 846. [Google Scholar] [CrossRef] [PubMed]
- Jegerlehner, A.; Schmitz, N.; Storni, T.; Bachmann, M.F. Influenza A vaccine based on the extracellular domain of M2: Weak protection mediated via antibody-dependent NK cell activity. J. Immunol. 2004, 172, 5598–5605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Bakkouri, K.; Descamps, F.; De Filette, M.; Smet, A.; Festjens, E.; Birkett, A.; Van Rooijen, N.; Verbeek, S.; Fiers, W.; Saelens, X. Universal vaccine based on ectodomain of matrix protein 2 of influenza A: Fc receptors and alveolar macrophages mediate protection. J. Immunol. 2011, 186, 1022–1031. [Google Scholar] [CrossRef] [Green Version]
- Yasui, F.; Kohara, M.; Kitabatake, M.; Nishiwaki, T.; Fujii, H.; Tateno, C.; Yoneda, M.; Morita, K.; Matsushima, K.; Koyasu, S.; et al. Phagocytic cells contribute to the antibody-mediated elimination of pulmonary-infected SARS coronavirus. Virology 2014, 454–455, 157–168. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, J.J.; Foltzer, M.; Chapman, S. The Fc portion of antibody to yellow fever virus NS1 is a determinant of protection against YF encephalitis in mice. Virology 1993, 192, 132–141. [Google Scholar] [CrossRef]
- Vogt, M.R.; Dowd, K.A.; Engle, M.; Tesh, R.B.; Johnson, S.; Pierson, T.C.; Diamond, M.S. Poorly neutralizing cross-reactive antibodies against the fusion loop of West Nile virus envelope protein protect in vivo via Fcgamma receptor and complement-dependent effector mechanisms. J. Virol. 2011, 85, 11567–11580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirkpatrick, E.; Henry, C.; McMahon, M.; Jiang, K.; Strohmeier, S.; van Bakel, H.; Wilson, P.C.; Krammer, F. Characterization of Novel Cross-Reactive Influenza B Virus Hemagglutinin Head Specific Antibodies That Lack Hemagglutination Inhibition Activity. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Asthagiri Arunkumar, G.; Ioannou, A.; Wohlbold, T.J.; Meade, P.; Aslam, S.; Amanat, F.; Ayllon, J.; Garcia-Sastre, A.; Krammer, F. Broadly Cross-Reactive, Nonneutralizing Antibodies against Influenza B Virus Hemagglutinin Demonstrate Effector Function-Dependent Protection against Lethal Viral Challenge in Mice. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Ko, E.J.; Kim, M.C.; Lee, Y.N.; Kim, K.H.; Jung, Y.J.; Kang, S.M. Roles of antibodies to influenza A virus hemagglutinin, neuraminidase, and M2e in conferring cross protection. Biochem. Biophys. Res. Commun. 2017, 493, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Yasuhara, A.; Yamayoshi, S.; Kiso, M.; Sakai-Tagawa, Y.; Koga, M.; Adachi, E.; Kikuchi, T.; Wang, I.H.; Yamada, S.; Kawaoka, Y. Antigenic drift originating from changes to the lateral surface of the neuraminidase head of influenza A virus. Nat. Microbiol. 2019, 4, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Bimler, L.; Ronzulli, S.L.; Song, A.Y.; Johnson, S.K.; Jones, C.A.; Kim, T.; Le, D.T.; Tompkins, S.M.; Paust, S. Matrix Protein 2 Extracellular Domain-Specific Monoclonal Antibodies Are an Effective and Potentially Universal Treatment for Influenza A. J. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.N.; Lee, Y.T.; Kim, M.C.; Hwang, H.S.; Lee, J.S.; Kim, K.H.; Kang, S.M. Fc receptor is not required for inducing antibodies but plays a critical role in conferring protection after influenza M2 vaccination. Immunology 2014, 143, 300–309. [Google Scholar] [CrossRef]
- Van den Hoecke, S.; Ehrhardt, K.; Kolpe, A.; El Bakkouri, K.; Deng, L.; Grootaert, H.; Schoonooghe, S.; Smet, A.; Bentahir, M.; Roose, K.; et al. Hierarchical and Redundant Roles of Activating FcgammaRs in Protection against Influenza Disease by M2e-Specific IgG1 and IgG2a Antibodies. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Grandea, A.G., 3rd; Olsen, O.A.; Cox, T.C.; Renshaw, M.; Hammond, P.W.; Chan-Hui, P.Y.; Mitcham, J.L.; Cieplak, W.; Stewart, S.M.; Grantham, M.L.; et al. Human antibodies reveal a protective epitope that is highly conserved among human and nonhuman influenza A viruses. Proc. Natl. Acad. Sci. USA 2010, 107, 12658–12663. [Google Scholar] [CrossRef] [Green Version]
- De Vlieger, D.; Hoffmann, K.; Van Molle, I.; Nerinckx, W.; Van Hoecke, L.; Ballegeer, M.; Creytens, S.; Remaut, H.; Hengel, H.; Schepens, B.; et al. Selective Engagement of FcgammaRIV by a M2e-Specific Single Domain Antibody Construct Protects Against Influenza A Virus Infection. Front. Immunol. 2019, 10, 2920. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Blanton, L.; Elal, A.I.A.; Alabi, N.; Barnes, J.; Biggerstaff, M.; Brammer, L.; Budd, A.P.; Burns, E.; Cummings, C.N.; et al. Update: Influenza Activity in the United States During the 2018–19 Season and Composition of the 2019–20 Influenza Vaccine. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 544–551. [Google Scholar] [CrossRef]
- Vigil, A.; Frias-Staheli, N.; Carabeo, T.; Wittekind, M. Airway Delivery of Anti-influenza Monoclonal Antibodies Results in Enhanced Antiviral Activities and Enables Broad-Coverage Combination Therapies. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Job, E.R.; Ysenbaert, T.; Smet, A.; Van Hecke, A.; Meuris, L.; Kleanthous, H.; Saelens, X.; Vogel, T.U. Fcgamma Receptors Contribute to the Antiviral Properties of Influenza Virus Neuraminidase-Specific Antibodies. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Corti, D.; Zhao, J.; Pedotti, M.; Simonelli, L.; Agnihothram, S.; Fett, C.; Fernandez-Rodriguez, B.; Foglierini, M.; Agatic, G.; Vanzetta, F.; et al. Prophylactic and postexposure efficacy of a potent human monoclonal antibody against MERS coronavirus. Proc. Natl. Acad. Sci. USA 2015, 112, 10473–10478. [Google Scholar] [CrossRef] [Green Version]
- Pascal, K.E.; Coleman, C.M.; Mujica, A.O.; Kamat, V.; Badithe, A.; Fairhurst, J.; Hunt, C.; Strein, J.; Berrebi, A.; Sisk, J.M.; et al. Pre- and postexposure efficacy of fully human antibodies against Spike protein in a novel humanized mouse model of MERS-CoV infection. Proc. Natl. Acad. Sci. USA 2015, 112, 8738–8743. [Google Scholar] [CrossRef] [Green Version]
- Luke, T.; Wu, H.; Zhao, J.; Channappanavar, R.; Coleman, C.M.; Jiao, J.A.; Matsushita, H.; Liu, Y.; Postnikova, E.N.; Ork, B.L.; et al. Human polyclonal immunoglobulin G from transchromosomic bovines inhibits MERS-CoV in vivo. Sci. Transl. Med. 2016, 8, 326ra21. [Google Scholar] [CrossRef] [Green Version]
- Qiu, H.; Sun, S.; Xiao, H.; Feng, J.; Guo, Y.; Tai, W.; Wang, Y.; Du, L.; Zhao, G.; Zhou, Y. Single-dose treatment with a humanized neutralizing antibody affords full protection of a human transgenic mouse model from lethal Middle East respiratory syndrome (MERS)-coronavirus infection. Antiviral. Res. 2016, 132, 141–148. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Zost, S.J.; Gilchuk, P.; Case, J.B.; Binshtein, E.; Chen, R.E.; Nkolola, J.P.; Schafer, A.; Reidy, J.X.; Trivette, A.; Nargi, R.S.; et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature 2020, 584, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Schafer, A.; Muecksch, F.; Lorenzi, J.C.C.; Leist, S.R.; Cipolla, M.; Bournazos, S.; Schmidt, F.; Gazumyan, A.; Baric, R.S.; Robbiani, D.F.; et al. Antibody potency, effector function and combinations in protection from SARS-CoV-2 infection in vivo. bioRxiv 2020. [Google Scholar] [CrossRef]
- Pinto, D.; Park, Y.J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; De Marco, A.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef]
- Liu, L.; Wei, Q.; Lin, Q.; Fang, J.; Wang, H.; Kwok, H.; Tang, H.; Nishiura, K.; Peng, J.; Tan, Z.; et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Chan, C.E.Z.; Seah, S.G.K.; Chye, D.H.; Massey, S.; Torres, M.; Lim, A.P.C.; Wong, S.K.K.; Neo, J.J.Y.; Wong, P.S.; Lim, J.H.; et al. The Fc-mediated effector functions of a potent SARS-CoV-2 neutralizing antibody, SC31, isolated from an early convalescent COVID-19 patient, are essential for the optimal therapeutic efficacy of the antibody. bioRxiv 2020. [Google Scholar] [CrossRef]
- Bournazos, S.; Gupta, A.; Ravetch, J.V. The role of IgG Fc receptors in antibody-dependent enhancement. Nat. Rev. Immunol. 2020, 20, 633–643. [Google Scholar] [CrossRef]
- Suryadevara, N.; Shrihari, S.; Gilchuk, P.; VanBlargan, L.A.; Binshtein, E.; Zost, S.J.; Nargi, R.S.; Sutton, R.E.; Winkler, E.S.; Chen, E.C.; et al. Neutralizing and protective human monoclonal antibodies recognizing the N-terminal domain of the SARS-CoV-2 spike protein. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lin, K.L.; Suzuki, Y.; Nakano, H.; Ramsburg, E.; Gunn, M.D. CCR2+ monocyte-derived dendritic cells and exudate macrophages produce influenza-induced pulmonary immune pathology and mortality. J. Immunol. 2008, 180, 2562–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kratofil, R.M.; Kubes, P.; Deniset, J.F. Monocyte Conversion during Inflammation and Injury. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microb. 2016, 19, 181–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tseng, C.T.; Sbrana, E.; Iwata-Yoshikawa, N.; Newman, P.C.; Garron, T.; Atmar, R.L.; Peters, C.J.; Couch, R.B. Immunization with SARS coronavirus vaccines leads to pulmonary immunopathology on challenge with the SARS virus. PLoS ONE 2012, 7, e35421. [Google Scholar] [CrossRef]
- Pedersen, N.C. An update on feline infectious peritonitis: Virology and immunopathogenesis. Vet. J. 2014, 201, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Lee, W.S.; Wheatley, A.K.; Kent, S.J.; DeKosky, B.J. Antibody-dependent enhancement and SARS-CoV-2 vaccines and therapies. Nat. Microbiol. 2020, 5, 1185–1191. [Google Scholar] [CrossRef]
- Clay, C.; Donart, N.; Fomukong, N.; Knight, J.B.; Lei, W.; Price, L.; Hahn, F.; Van Westrienen, J.; Harrod, K.S. Primary severe acute respiratory syndrome coronavirus infection limits replication but not lung inflammation upon homologous rechallenge. J. Virol. 2012, 86, 4234–4244. [Google Scholar] [CrossRef] [Green Version]
- Bolles, M.; Deming, D.; Long, K.; Agnihothram, S.; Whitmore, A.; Ferris, M.; Funkhouser, W.; Gralinski, L.; Totura, A.; Heise, M.; et al. A double-inactivated severe acute respiratory syndrome coronavirus vaccine provides incomplete protection in mice and induces increased eosinophilic proinflammatory pulmonary response upon challenge. J. Virol. 2011, 85, 12201–12215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, R.; Shan, C.; Duan, X.; Chen, Z.; Liu, P.; Song, J.; Song, T.; Bi, X.; Han, C.; Wu, L.; et al. A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2. Nature 2020, 584, 120–124. [Google Scholar] [CrossRef]
- Larsen, M.D.; de Graaf, E.L.; Sonneveld, M.E.; Plomp, H.R.; Nouta, J.; Hoepel, W.; Chen, H.J.; Linty, F.; Visser, R.; Brinkhaus, M.; et al. Afucosylated IgG characterizes enveloped viral responses and correlates with COVID-19 severity. Science 2021, 371, eabc8378. [Google Scholar] [CrossRef]
- Atyeo, C.; Fischinger, S.; Zohar, T.; Slein, M.D.; Burke, J.; Loos, C.; McCulloch, D.J.; Newman, K.L.; Wolf, C.; Yu, J.; et al. Distinct Early Serological Signatures Track with SARS-CoV-2 Survival. Immunity 2020, 53, 524–532.e4. [Google Scholar] [CrossRef]
- Ryman, K.D.; Klimstra, W.B. Host responses to alphavirus infection. Immunol. Rev. 2008, 225, 27–45. [Google Scholar] [CrossRef]
- Cunha, M.S.; Costa, P.A.G.; Correa, I.A.; de Souza, M.R.M.; Calil, P.T.; da Silva, G.P.D.; Costa, S.M.; Fonseca, V.W.P.; da Costa, L.J. Chikungunya Virus: An Emergent Arbovirus to the South American Continent and a Continuous Threat to the World. Front. Microbiol. 2020, 11, 1297. [Google Scholar] [CrossRef]
- Guzman-Teran, C.; Calderon-Rangel, A.; Rodriguez-Morales, A.; Mattar, S. Venezuelan equine encephalitis virus: The problem is not over for tropical America. Ann. Clin. Microbiol. Antimicrob. 2020, 19, 19. [Google Scholar] [CrossRef]
- Lescar, J.; Roussel, A.; Wien, M.W.; Navaza, J.; Fuller, S.D.; Wengler, G.; Wengler, G.; Rey, F.A. The Fusion glycoprotein shell of Semliki Forest virus: An icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 2001, 105, 137–148. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef]
- Sun, S.; Xiang, Y.; Akahata, W.; Holdaway, H.; Pal, P.; Zhang, X.; Diamond, M.S.; Nabel, G.J.; Rossmann, M.G. Structural analyses at pseudo atomic resolution of Chikungunya virus and antibodies show mechanisms of neutralization. eLife 2013, 2, e00435. [Google Scholar] [CrossRef]
- Voss, J.E.; Vaney, M.C.; Duquerroy, S.; Vonrhein, C.; Girard-Blanc, C.; Crublet, E.; Thompson, A.; Bricogne, G.; Rey, F.A. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 2010, 468, 709–712. [Google Scholar] [CrossRef]
- Fox, J.M.; Long, F.; Edeling, M.A.; Lin, H.; van Duijl-Richter, M.K.S.; Fong, R.H.; Kahle, K.M.; Smit, J.M.; Jin, J.; Simmons, G.; et al. Broadly Neutralizing Alphavirus Antibodies Bind an Epitope on E2 and Inhibit Entry and Egress. Cell 2015, 163, 1095–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, P.; Dowd, K.A.; Brien, J.D.; Edeling, M.A.; Gorlatov, S.; Johnson, S.; Lee, I.; Akahata, W.; Nabel, G.J.; Richter, M.K.; et al. Development of a highly protective combination monoclonal antibody therapy against Chikungunya virus. PLoS Pathog. 2013, 9, e1003312. [Google Scholar] [CrossRef] [Green Version]
- Jin, J.; Galaz-Montoya, J.G.; Sherman, M.B.; Sun, S.Y.; Goldsmith, C.S.; O’Toole, E.T.; Ackerman, L.; Carlson, L.A.; Weaver, S.C.; Chiu, W.; et al. Neutralizing Antibodies Inhibit Chikungunya Virus Budding at the Plasma Membrane. Cell Host Microbe 2018, 24, 417–428.e5. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.F.; Fox, J.M.; Earnest, J.T.; Ng, T.S.; Kim, A.S.; Fibriansah, G.; Kostyuchenko, V.A.; Shi, J.; Shu, B.; Diamond, M.S.; et al. Structural basis of Chikungunya virus inhibition by monoclonal antibodies. Proc. Natl. Acad. Sci. USA 2020, 117, 27637–27645. [Google Scholar] [CrossRef]
- Earnest, J.T.; Basore, K.; Roy, V.; Bailey, A.L.; Wang, D.; Alter, G.; Fremont, D.H.; Diamond, M.S. Neutralizing antibodies against Mayaro virus require Fc effector functions for protective activity. J. Exp. Med. 2019, 216, 2282–2301. [Google Scholar] [CrossRef]
- Fox, J.M.; Huang, L.; Tahan, S.; Powell, L.A.; Crowe, J.E., Jr.; Wang, D.; Diamond, M.S. A cross-reactive antibody protects against Ross River virus musculoskeletal disease despite rapid neutralization escape in mice. PLoS Pathog. 2020, 16, e1008743. [Google Scholar] [CrossRef] [PubMed]
- Powell, L.A.; Miller, A.; Fox, J.M.; Kose, N.; Klose, T.; Kim, A.S.; Bombardi, R.; Tennekoon, R.N.; Dharshan de Silva, A.; Carnahan, R.H.; et al. Human mAbs Broadly Protect against Arthritogenic Alphaviruses by Recognizing Conserved Elements of the Mxra8 Receptor-Binding Site. Cell Host Microbe 2020, 28, 699–711.e7. [Google Scholar] [CrossRef]
- Rulli, N.E.; Suhrbier, A.; Hueston, L.; Heise, M.T.; Tupanceska, D.; Zaid, A.; Wilmes, A.; Gilmore, K.; Lidbury, B.A.; Mahalingam, S. Ross River virus: Molecular and cellular aspects of disease pathogenesis. Pharmacol. Ther. 2005, 107, 329–342. [Google Scholar] [CrossRef]
- Linn, M.L.; Aaskov, J.G.; Suhrbier, A. Antibody-dependent enhancement and persistence in macrophages of an arbovirus associated with arthritis. J. Gen. Virol. 1996, 77, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Lidbury, B.A.; Mahalingam, S. Specific ablation of antiviral gene expression in macrophages by antibody-dependent enhancement of Ross River virus infection. J. Virol. 2000, 74, 8376–8381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lum, F.M.; Couderc, T.; Chia, B.S.; Ong, R.Y.; Her, Z.; Chow, A.; Leo, Y.S.; Kam, Y.W.; Renia, L.; Lecuit, M.; et al. Antibody-mediated enhancement aggravates chikungunya virus infection and disease severity. Sci. Rep. 2018, 8, 1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, A.S.; Austin, S.K.; Gardner, C.L.; Zuiani, A.; Reed, D.S.; Trobaugh, D.W.; Sun, C.; Basore, K.; Williamson, L.E.; Crowe, J.E., Jr.; et al. Protective antibodies against Eastern equine encephalitis virus bind to epitopes in domains A and B of the E2 glycoprotein. Nat. Microbiol. 2019, 4, 187–197. [Google Scholar] [CrossRef]
- Burke, C.W.; Froude, J.W.; Miethe, S.; Hulseweh, B.; Hust, M.; Glass, P.J. Human-Like Neutralizing Antibodies Protect Mice from Aerosol Exposure with Western Equine Encephalitis Virus. Viruses 2018, 10, 147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, A.R.; Bowen, R.A.; Frederickson, S.; Maruyama, T.; Roehrig, J.T.; Blair, C.D. Treatment of mice with human monoclonal antibody 24h after lethal aerosol challenge with virulent Venezuelan equine encephalitis virus prevents disease but not infection. Virology 2011, 414, 146–152. [Google Scholar] [CrossRef] [Green Version]
- Burke, C.W.; Froude, J.W.; Rossi, F.; White, C.E.; Moyer, C.L.; Ennis, J.; Pitt, M.L.; Streatfield, S.; Jones, R.M.; Musiychuk, K.; et al. Therapeutic monoclonal antibody treatment protects nonhuman primates from severe Venezuelan equine encephalitis virus disease after aerosol exposure. PLoS Pathog. 2019, 15, e1008157. [Google Scholar] [CrossRef] [Green Version]
- Williamson, L.E.; Gilliland, T., Jr.; Yadav, P.K.; Binshtein, E.; Bombardi, R.; Kose, N.; Nargi, R.S.; Sutton, R.E.; Durie, C.L.; Armstrong, E.; et al. Human Antibodies Protect against Aerosolized Eastern Equine Encephalitis Virus Infection. Cell 2020, 183, 1884–1900.e23. [Google Scholar] [CrossRef]
- Chong, H.Y.; Leow, C.Y.; Abdul Majeed, A.B.; Leow, C.H. Flavivirus infection-A review of immunopathogenesis, immunological response, and immunodiagnosis. Virus Res. 2019, 274, 197770. [Google Scholar] [CrossRef]
- Reyes-Sandoval, A.; Ludert, J.E. The Dual Role of the Antibody Response Against the Flavivirus Non-structural Protein 1 (NS1) in Protection and Immuno-Pathogenesis. Front. Immunol. 2019, 10, 1651. [Google Scholar] [CrossRef] [Green Version]
- Chung, K.M.; Liszewski, M.K.; Nybakken, G.; Davis, A.E.; Townsend, R.R.; Fremont, D.H.; Atkinson, J.P.; Diamond, M.S. West Nile virus nonstructural protein NS1 inhibits complement activation by binding the regulatory protein factor H. Proc. Natl. Acad. Sci. USA 2006, 103, 19111–19116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Modhiran, N.; Watterson, D.; Muller, D.A.; Panetta, A.K.; Sester, D.P.; Liu, L.; Hume, D.A.; Stacey, K.J.; Young, P.R. Dengue virus NS1 protein activates cells via Toll-like receptor 4 and disrupts endothelial cell monolayer integrity. Sci. Transl. Med. 2015, 7, 304ra142. [Google Scholar] [CrossRef] [Green Version]
- Watterson, D.; Modhiran, N.; Young, P.R. The many faces of the flavivirus NS1 protein offer a multitude of options for inhibitor design. Antiviral. Res. 2016, 130, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.J.; Duehr, J.; Dulin, H.; Broecker, F.; Brown, J.A.; Arumemi, F.O.; Bermudez Gonzalez, M.C.; Leyva-Grado, V.H.; Evans, M.J.; Simon, V.; et al. Human antibodies targeting Zika virus NS1 provide protection against disease in a mouse model. Nat. Commun. 2018, 9, 4560. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Liu, X.; Ye, X.; Su, W.; Zhang, X.; Deng, W.; Luo, J.; Xiang, M.; Guo, W.; Zhang, S.; et al. Monoclonal Antibodies against Zika Virus NS1 Protein Confer Protection via Fcgamma Receptor-Dependent and -Independent Pathways. mBio 2021, 12. [Google Scholar] [CrossRef]
- Brault, A.C.; Domi, A.; McDonald, E.M.; Talmi-Frank, D.; McCurley, N.; Basu, R.; Robinson, H.L.; Hellerstein, M.; Duggal, N.K.; Bowen, R.A.; et al. A Zika Vaccine Targeting NS1 Protein Protects Immunocompetent Adult Mice in a Lethal Challenge Model. Sci. Rep. 2017, 7, 14769. [Google Scholar] [CrossRef]
- Bailey, M.J.; Broecker, F.; Duehr, J.; Arumemi, F.; Krammer, F.; Palese, P.; Tan, G.S. Antibodies Elicited by an NS1-Based Vaccine Protect Mice against Zika Virus. mBio 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, J.J.; Brandriss, M.W.; Walsh, E.E. Protection against 17D yellow fever encephalitis in mice by passive transfer of monoclonal antibodies to the nonstructural glycoprotein gp48 and by active immunization with gp48. J. Immunol. 1985, 135, 2805–2809. [Google Scholar]
- Chung, K.M.; Nybakken, G.E.; Thompson, B.S.; Engle, M.J.; Marri, A.; Fremont, D.H.; Diamond, M.S. Antibodies against West Nile Virus nonstructural protein NS1 prevent lethal infection through Fc gamma receptor-dependent and -independent mechanisms. J. Virol. 2006, 80, 1340–1351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, K.M.; Thompson, B.S.; Fremont, D.H.; Diamond, M.S. Antibody recognition of cell surface-associated NS1 triggers Fc-gamma receptor-mediated phagocytosis and clearance of West Nile Virus-infected cells. J. Virol. 2007, 81, 9551–9555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biering, S.B.; Akey, D.L.; Wong, M.P.; Brown, W.C.; Lo, N.T.N.; Puerta-Guardo, H.; Tramontini Gomes de Sousa, F.; Wang, C.; Konwerski, J.R.; Espinosa, D.A.; et al. Structural basis for antibody inhibition of flavivirus NS1-triggered endothelial dysfunction. Science 2021, 371, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Modhiran, N.; Song, H.; Liu, L.; Bletchly, C.; Brillault, L.; Amarilla, A.A.; Xu, X.; Qi, J.; Chai, Y.; Cheung, S.T.M.; et al. A broadly protective antibody that targets the flavivirus NS1 protein. Science 2021, 371, 190–194. [Google Scholar] [CrossRef]
- Chu, Y.T.; Wan, S.W.; Chang, Y.C.; Lee, C.K.; Wu-Hsieh, B.A.; Anderson, R.; Lin, Y.S. Antibodies against nonstructural protein 1 protect mice from dengue virus-induced mast cell activation. Lab. Investig. 2017, 97, 602–614. [Google Scholar] [CrossRef] [Green Version]
- Amarasinghe, G.K.; Bao, Y.; Basler, C.F.; Bavari, S.; Beer, M.; Bejerman, N.; Blasdell, K.R.; Bochnowski, A.; Briese, T.; Bukreyev, A.; et al. Taxonomy of the order Mononegavirales: Update 2017. Arch. Virol. 2017, 162, 2493–2504. [Google Scholar] [CrossRef] [PubMed]
- Takada, A. Filovirus tropism: Cellular molecules for viral entry. Front. Microbiol. 2012, 3, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manicassamy, B.; Wang, J.; Rumschlag, E.; Tymen, S.; Volchkova, V.; Volchkov, V.; Rong, L. Characterization of Marburg virus glycoprotein in viral entry. Virology 2007, 358, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.Y.; Duckers, H.J.; Sullivan, N.J.; Sanchez, A.; Nabel, E.G.; Nabel, G.J. Identification of the Ebola virus glycoprotein as the main viral determinant of vascular cell cytotoxicity and injury. Nat. Med. 2000, 6, 886–889. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Misasi, J.; Mulangu, S.; Stanley, D.A.; Kanekiyo, M.; Wollen, S.; Ploquin, A.; Doria-Rose, N.A.; Staupe, R.P.; Bailey, M.; et al. Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science 2016, 351, 1339–1342. [Google Scholar] [CrossRef] [Green Version]
- Wec, A.Z.; Herbert, A.S.; Murin, C.D.; Nyakatura, E.K.; Abelson, D.M.; Fels, J.M.; He, S.; James, R.M.; de La Vega, M.A.; Zhu, W.; et al. Antibodies from a Human Survivor Define Sites of Vulnerability for Broad Protection against Ebolaviruses. Cell 2017, 169, 878–890.e15. [Google Scholar] [CrossRef]
- Wec, A.Z.; Bornholdt, Z.A.; He, S.; Herbert, A.S.; Goodwin, E.; Wirchnianski, A.S.; Gunn, B.M.; Zhang, Z.; Zhu, W.; Liu, G.; et al. Development of a Human Antibody Cocktail that Deploys Multiple Functions to Confer Pan-Ebolavirus Protection. Cell Host Microbe 2019, 25, 39–48.e5. [Google Scholar] [CrossRef] [Green Version]
- Bornholdt, Z.A.; Turner, H.L.; Murin, C.D.; Li, W.; Sok, D.; Souders, C.A.; Piper, A.E.; Goff, A.; Shamblin, J.D.; Wollen, S.E.; et al. Isolation of potent neutralizing antibodies from a survivor of the 2014 Ebola virus outbreak. Science 2016, 351, 1078–1083. [Google Scholar] [CrossRef] [Green Version]
- Gunn, B.M.; Yu, W.H.; Karim, M.M.; Brannan, J.M.; Herbert, A.S.; Wec, A.Z.; Halfmann, P.J.; Fusco, M.L.; Schendel, S.L.; Gangavarapu, K.; et al. A Role for Fc Function in Therapeutic Monoclonal Antibody-Mediated Protection against Ebola Virus. Cell Host Microbe 2018, 24, 221–233.e5. [Google Scholar] [CrossRef] [PubMed]
- Saphire, E.O.; Schendel, S.L.; Fusco, M.L.; Gangavarapu, K.; Gunn, B.M.; Wec, A.Z.; Halfmann, P.J.; Brannan, J.M.; Herbert, A.S.; Qiu, X.; et al. Systematic Analysis of Monoclonal Antibodies against Ebola Virus GP Defines Features that Contribute to Protection. Cell 2018, 174, 938–952.e13. [Google Scholar] [CrossRef] [Green Version]
- Qiu, X.; Audet, J.; Wong, G.; Fernando, L.; Bello, A.; Pillet, S.; Alimonti, J.B.; Kobinger, G.P. Sustained protection against Ebola virus infection following treatment of infected nonhuman primates with ZMAb. Sci. Rep. 2013, 3, 3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettitt, J.; Zeitlin, L.; Kim, D.H.; Working, C.; Johnson, J.C.; Bohorov, O.; Bratcher, B.; Hiatt, E.; Hume, S.D.; Johnson, A.K.; et al. Therapeutic intervention of Ebola virus infection in rhesus macaques with the MB-003 monoclonal antibody cocktail. Sci. Transl. Med. 2013, 5, 199ra113. [Google Scholar] [CrossRef] [Green Version]
- Olinger, G.G., Jr.; Pettitt, J.; Kim, D.; Working, C.; Bohorov, O.; Bratcher, B.; Hiatt, E.; Hume, S.D.; Johnson, A.K.; Morton, J.; et al. Delayed treatment of Ebola virus infection with plant-derived monoclonal antibodies provides protection in rhesus macaques. Proc. Natl. Acad. Sci. USA 2012, 109, 18030–18035. [Google Scholar] [CrossRef] [Green Version]
- Oswald, W.B.; Geisbert, T.W.; Davis, K.J.; Geisbert, J.B.; Sullivan, N.J.; Jahrling, P.B.; Parren, P.W.; Burton, D.R. Neutralizing antibody fails to impact the course of Ebola virus infection in monkeys. PLoS Pathog. 2007, 3, e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, J.A.; Hevey, M.; Bakken, R.; Guest, S.; Bray, M.; Schmaljohn, A.L.; Hart, M.K. Epitopes involved in antibody-mediated protection from Ebola virus. Science 2000, 287, 1664–1666. [Google Scholar] [CrossRef]
- Ilinykh, P.A.; Santos, R.I.; Gunn, B.M.; Kuzmina, N.A.; Shen, X.; Huang, K.; Gilchuk, P.; Flyak, A.I.; Younan, P.; Alter, G.; et al. Asymmetric antiviral effects of ebolavirus antibodies targeting glycoprotein stem and glycan cap. PLoS Pathog. 2018, 14, e1007204. [Google Scholar] [CrossRef]
- Holtsberg, F.W.; Shulenin, S.; Vu, H.; Howell, K.A.; Patel, S.J.; Gunn, B.; Karim, M.; Lai, J.R.; Frei, J.C.; Nyakatura, E.K.; et al. Pan-ebolavirus and Pan-filovirus Mouse Monoclonal Antibodies: Protection against Ebola and Sudan Viruses. J. Virol. 2016, 90, 266–278. [Google Scholar] [CrossRef] [Green Version]
- Ilinykh, P.A.; Huang, K.; Santos, R.I.; Gilchuk, P.; Gunn, B.M.; Karim, M.M.; Liang, J.; Fouch, M.E.; Davidson, E.; Parekh, D.V.; et al. Non-neutralizing Antibodies from a Marburg Infection Survivor Mediate Protection by Fc-Effector Functions and by Enhancing Efficacy of Other Antibodies. Cell Host Microbe 2020, 27, 976–991.e11. [Google Scholar] [CrossRef]
- Markham, A. REGN-EB3: First Approval. Drugs 2021, 81, 175–178. [Google Scholar] [CrossRef]
- Deb, P.; Molla, M.M.A.; Rahman, K.M.S. An update to monoclonal antibody as therapeutic option against COVID-19. Biosaf. Health 2021, 3, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Sterlin, D.; Gorochov, G. When Therapeutic IgA Antibodies Might Come of Age. Pharmacology 2021, 106, 9–19. [Google Scholar] [CrossRef]
- Marovich, M.; Mascola, J.R.; Cohen, M.S. Monoclonal Antibodies for Prevention and Treatment of COVID-19. JAMA 2020, 324, 131–132. [Google Scholar] [CrossRef] [PubMed]
- Castro-Dopico, T.; Clatworthy, M.R. IgG and Fcγ Receptors in Intestinal Immunity and Inflammation. Front. Immunol. 2019, 10, 805. [Google Scholar] [CrossRef] [PubMed]
- Menikou, S.; Langford, P.R.; Levin, M. Kawasaki Disease: The Role of Immune Complexes Revisited. Front. Immunol. 2019, 10, 1156. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Mouse FcγR | Function | Mouse IgG Interactions | Human IgG Interactions | Cellular Expression in Mice |
|---|---|---|---|---|
| FcγRI | Activation | IgG2a/c IgG2b IgG3 # | IgG1 IgG3 IgG4 | dendritic cells macrophages monocytes |
| FcγRIIb | Inhibition | IgG1 IgG2b IgG2a/c | IgG1 IgG2 IgG3 IgG4 | neutrophils B cells dendritic cells macrophages monocytes |
| FcγRIII | Activation | IgG1 IgG2a/c IgG2b | IgG1 IgG2 IgG3 IgG4 | NK cells neutrophils dendritic cells macrophages monocytes |
| FcγRIV | Activation | IgG2a/c IgG2b | IgG1 IgG3 IgG4 * | neutrophils dendritic cells macrophages monocytes |
| Virus | Antibody (Species) | Neut + | Identified Cell Type (Depletion or Transfer Method) | Outcome $ | Ref ** |
|---|---|---|---|---|---|
| Influenza A virus | 2B9, 2C10, FEE8 (mouse) | No | AMΦ * [clodronate liposomes or adoptive transfer of AMΦ (2B9)] | Depletion- increased weight loss, viral load at 6 dpi, and mortality | [56] |
| Transfer- reduced weight loss and increased survival | |||||
| 5E01, 5D06 (human) | No | AMΦ (clodronate liposomes) | Depletion- increased weight loss, mortality | [56] | |
| 9H10, 6F12 (mouse) | Yes | AMΦ (clodronate liposomes) | Depletion with suboptimal mAb dose- increased weight loss | [56] | |
| FI6v3 (optimized human) | Yes | CD8+ T cells (anti-CD8) | GAALIE or GA variant plus depletion- increased weight loss and mortality. | [35] | |
| M2e-HBc immunized | No | NK and NKT cells (anti-asialo-GM1) | Depletion- increased mortality | [57] | |
| M2e-HBc immune serum (passive transfer) | No | AMΦ * (clodronate liposomes or adoptive transfer of AMΦ) | Depletion- increased weight loss and mortality | [58] | |
| Transfer- reduced weight loss and increased survival | |||||
| SARS-CoV-2 | COV2-2050 (human) | Yes | Ly6Chi monocytes (anti-CCR2) | Depletion- increased weight loss, lung pathology, and cytokines and chemokines | [11] |
| CD8+ T cells (anti-CD8) | Depletion- increased viral burden at 8dpi in lungs | ||||
| SARS-CoV | Immune serum | Yes | AMΦ and monocytes (clodronate liposomes and/or anti-Gr1) | Depletion- increased viral titer in lungs at 9 dpi. Highest titer when both AMΦ and monocytes were depleted | [59] |
| Chikungunya virus | CHK-152 + CHK-166 (mouse & humanized) | Yes | Monocytes (anti-CCR2) | Depletion- increased viral burden in ankle at 7 dpi and increased foot swelling | [12] |
| Yellow Fever virus | 1A5 | No | “killer cells” (cyclophosphamide treatment) | Depletion- reduced survival | [60] |
| West Nile virus | E28 | No | Macrophages (clodronate liposomes) | Depletion- reduced viremia 1 and 2 dpi | [61] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keeler, S.P.; Fox, J.M. Requirement of Fc-Fc Gamma Receptor Interaction for Antibody-Based Protection against Emerging Virus Infections. Viruses 2021, 13, 1037. https://doi.org/10.3390/v13061037
Keeler SP, Fox JM. Requirement of Fc-Fc Gamma Receptor Interaction for Antibody-Based Protection against Emerging Virus Infections. Viruses. 2021; 13(6):1037. https://doi.org/10.3390/v13061037
Chicago/Turabian StyleKeeler, Shamus P., and Julie M. Fox. 2021. "Requirement of Fc-Fc Gamma Receptor Interaction for Antibody-Based Protection against Emerging Virus Infections" Viruses 13, no. 6: 1037. https://doi.org/10.3390/v13061037
APA StyleKeeler, S. P., & Fox, J. M. (2021). Requirement of Fc-Fc Gamma Receptor Interaction for Antibody-Based Protection against Emerging Virus Infections. Viruses, 13(6), 1037. https://doi.org/10.3390/v13061037