
The Potential of Phage Therapy against the Emerging Opportunistic Pathogen Stenotrophomonas maltophilia



Abstract
:1. Introduction
2. Stenotrophomonas maltophilia
2.1. Clinical Prevalence and Significance
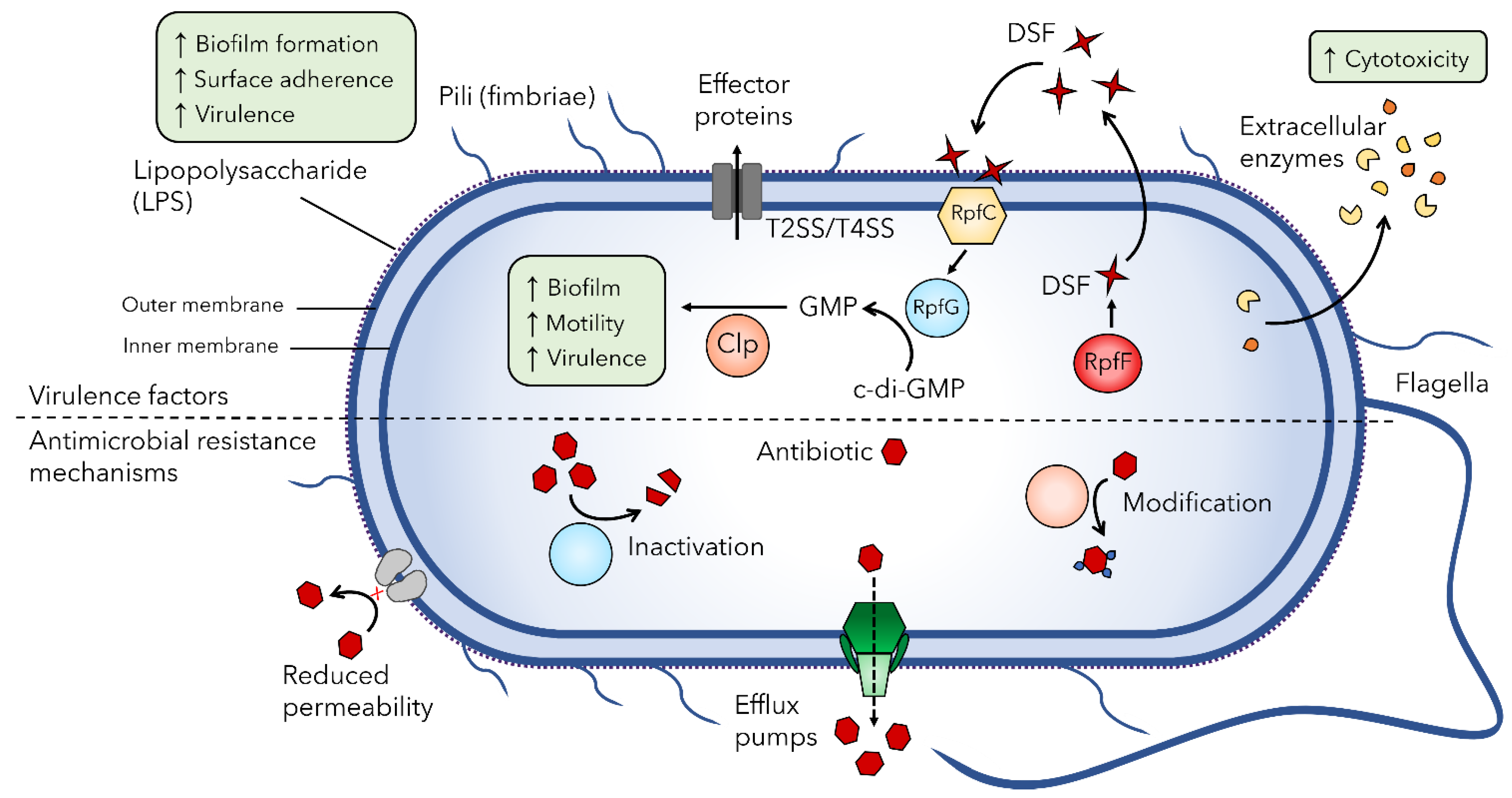
2.2. S. maltophilia Virulence Factors
2.3. Antimicrobial Resistance Mechanisms of S. maltophilia
3. Phage Therapy
3.1. Bacteriophages
3.2. Clinical Data Using Phages
3.3. Phage Therapy Strategies
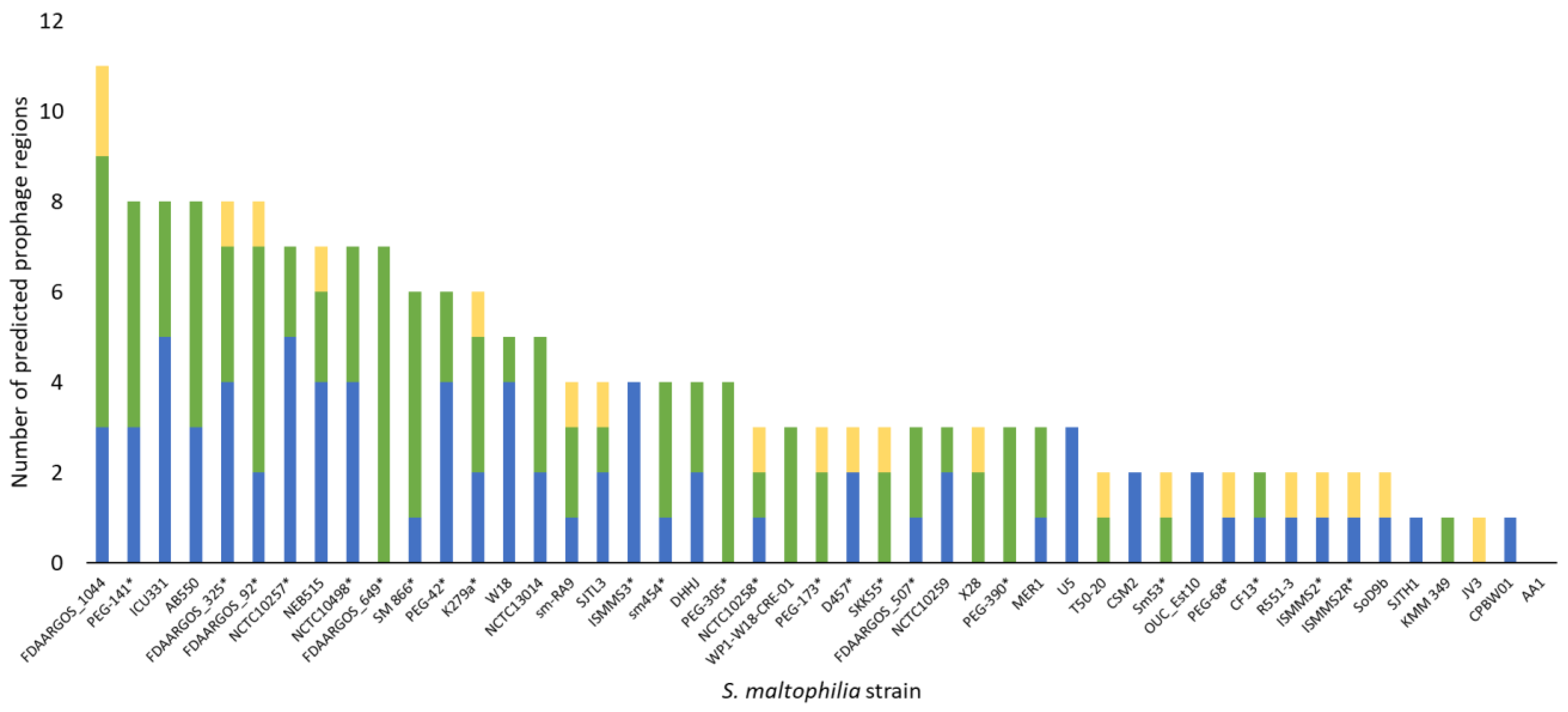
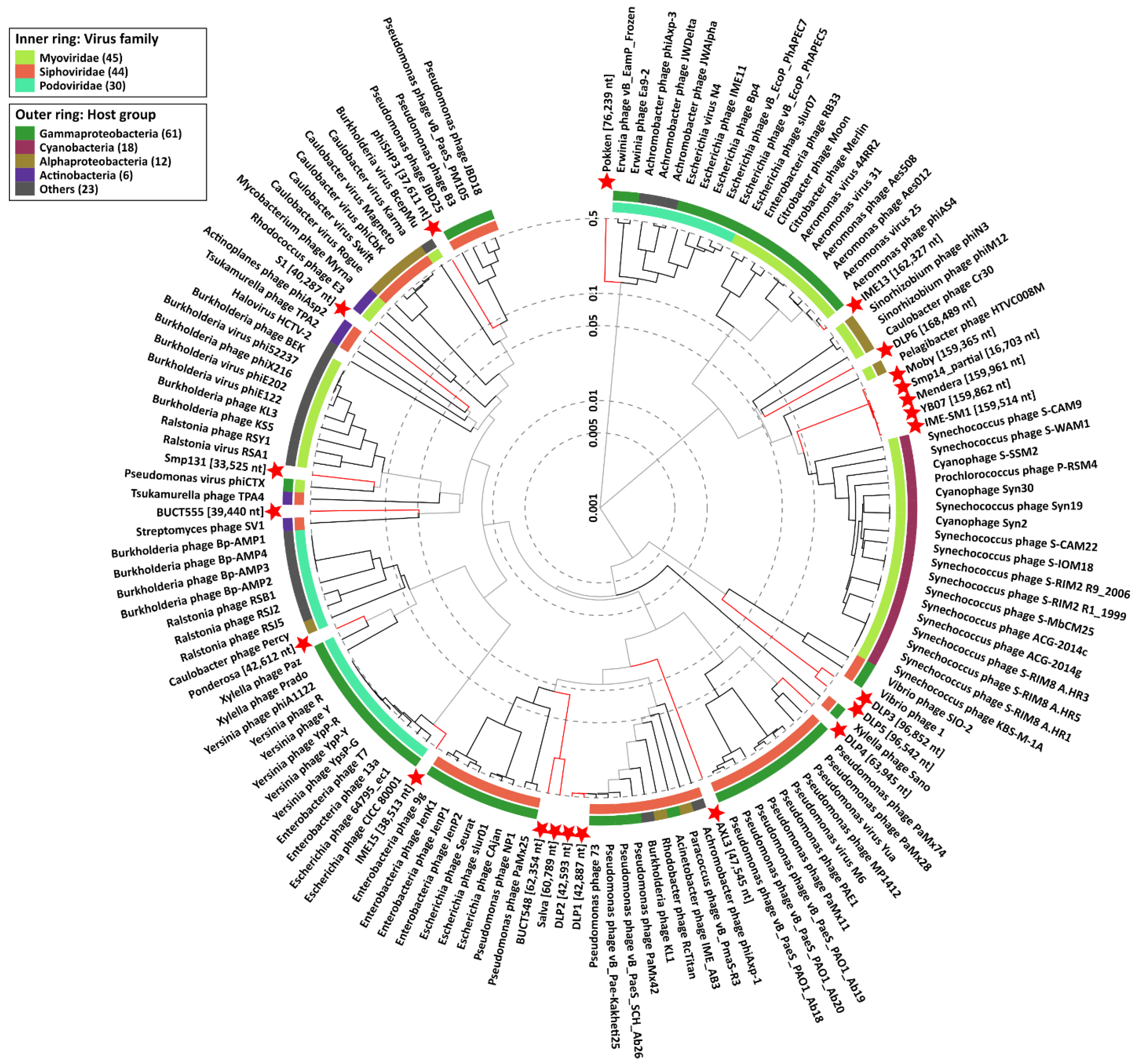
3.4. S. maltophilia Phages
3.5. Potential of Phage Therapy for S. maltophilia
3.6. Perspectives and Future Directions
4. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- O’Neill, J. Tackling drug-resistant infections globally: Final report and recommendations. Rev. Antimicrob. Resist. 2016, 1–84. Available online: https://amr-review.org/sites/default/files/160518_Final%20paper_with%20cover.pdf (accessed on 7 May 2018).
- When Antibiotics Fail: The Expert Panel on the Potential Socio-Economic Impacts of Antimicrobial Resistance in Canada; Council of Canadian Academies: Ottawa, ON, Canada, 2019; ISBN 9781926522753.
- Brooke, J.S. Stenotrophomonas maltophilia: An emerging global opportunistic pathogen. Clin. Microbiol. Rev. 2012, 25, 2–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.P.; Lee Wong, A.C. Extracellular fatty acids facilitate flagella-independent translocation by Stenotrophomonas maltophilia. Res. Microbiol. 2007, 158, 702–711. [Google Scholar] [CrossRef] [PubMed]
- Pompilio, A.; Pomponio, S.; Crocetta, V.; Gherardi, G.; Verginelli, F.; Fiscarelli, E.; Dicuonzo, G.; Savini, V.; D’Antonio, D.; Di Bonaventura, G. Phenotypic and genotypic characterization of Stenotrophomonas maltophilia isolates from patients with cystic fibrosis: Genome diversity, biofilm formation, and virulence. BMC Microbiol. 2011, 11, 159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, R.P.; Monchy, S.; Cardinale, M.; Taghavi, S.; Crossman, L.; Avison, M.B.; Berg, G.; van der Lelie, D.; Dow, J.M. The versatility and adaptation of bacteria from the genus Stenotrophomonas. Nat. Rev. Microbiol. 2009, 7, 514–525. [Google Scholar] [CrossRef]
- Hugh, R.; Ryschenkow, E. Pseudomonas maltophilia, an Alcaligenes-like species. J. Gen. Microbiol. 1961, 26, 123–132. [Google Scholar] [CrossRef] [Green Version]
- Swings, J.; De Vos, P.; Van den Mooter, M.; De Ley, J. Transfer of Pseudomonas maltophilia Hugh 1981 to the genus Xanthomonas as Xanthomonas maltophilia (Hugh 1981) comb. nov. Int. J. Syst. Bacteriol. 1983, 33, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Palleroni, N.J.; Bradbury, J.F. Stenotrophomonas, a new bacterial genus for Xanthomonas maltophilia (Hugh 1980) Swings et al. 1983. Int. J. Syst. Bacteriol. 1993, 43, 606–609. [Google Scholar] [CrossRef] [Green Version]
- Svensson-Stadler, L.A.; Mihaylova, S.A.; Moore, E.R.B. Stenotrophomonas interspecies differentiation and identification by gyrB sequence analysis. FEMS Microbiol. Lett. 2012, 327, 15–24. [Google Scholar] [CrossRef] [Green Version]
- Peters, D.L.; Lynch, K.H.; Stothard, P.; Dennis, J.J. The isolation and characterization of two Stenotrophomonas maltophilia bacteriophages capable of cross-taxonomic order infectivity. BMC Genom. 2015, 16, 664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berg, G.; Roskot, N.; Smalla, K. Genotypic and phenotypic relationships between clinical and environmental isolates of Stenotrophomonas maltophilia. J. Clin. Microbiol. 1999, 37, 3594–3600. [Google Scholar] [CrossRef] [Green Version]
- Berg, G.; Martinez, J.L. Friends or foes: Can we make a distinction between beneficial and harmful strains of the Stenotrophomonas maltophilia complex? Front. Microbiol. 2015, 6, 241. [Google Scholar] [CrossRef]
- Pages, D.; Rose, J.; Conrod, S.; Cuine, S.; Carrier, P.; Heulin, T.; Achouak, W. Heavy metal tolerance in Stenotrophomonas maltophilia. PLoS ONE 2008, 3, e1539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mukherjee, P.; Roy, P. Genomic potential of Stenotrophomonas maltophilia in bioremediation with an assessment of its multifaceted role in our environment. Front. Microbiol. 2016, 7, 967. [Google Scholar] [CrossRef]
- Pompilio, A.; Crocetta, V.; Ghosh, D.; Chakrabarti, M.; Gherardi, G.; Vitali, L.A.; Fiscarelli, E.; Di Bonaventura, G. Stenotrophomonas maltophilia phenotypic and genotypic diversity during a 10-year colonization in the lungs of a cystic fibrosis patient. Front. Microbiol. 2016, 7, 1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdezate, S.; Vindel, A.; Martín-Dávila, P.; Del Saz, B.S.; Baquero, F.; Cantón, R. High genetic diversity among Stenotrophomonas maltophilia strains despite their originating at a single hospital. J. Clin. Microbiol. 2004, 42, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turrientes, M.C.; Baquero, M.R.; Sánchez, M.B.; Valdezate, S.; Escudero, E.; Berg, G.; Cantón, R.; Baquero, F.; Galán, J.C.; Martínez, J.L. Polymorphic mutation frequencies of clinical and environmental Stenotrophomonas maltophilia populations. Appl. Environ. Microbiol. 2010, 76, 1746–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.T.; Lin, C.Y.; Chen, Y.H.; Hsueh, P.R. Update on infections caused by Stenotrophomonas maltophilia with particular attention to resistance mechanisms and therapeutic options. Front. Microbiol. 2015, 6, 893. [Google Scholar] [CrossRef]
- Canadian Antimicrobial Resistance Alliance CANWARD Pathogens. 2018. Available online: http://www.can-r.com/study.php?study=canw2018&year=2018 (accessed on 26 April 2021).
- Gröschel, M.I.; Meehan, C.J.; Barilar, I.; Diricks, M.; Gonzaga, A.; Steglich, M.; Conchillo-Solé, O.; Scherer, I.C.; Mamat, U.; Luz, C.F.; et al. The phylogenetic landscape and nosocomial spread of the multidrug-resistant opportunist Stenotrophomonas maltophilia. Nat. Commun. 2020, 11, 28. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization Public Health Importance of Antimicrobial Resistance. Available online: https://www.who.int/drugresistance/AMR_Importance/en/ (accessed on 26 April 2021).
- Al-Anazi, K.A.; Al-Jasser, A.M. Infections caused by Stenotrophomonas maltophilia in recipients of hematopoietic stem cell transplantation. Front. Oncol. 2014, 4, 232. [Google Scholar] [CrossRef] [Green Version]
- Silbaq, F.S. Viable ultramicrocells in drinking water. J. Appl. Microbiol. 2009, 106, 106–117. [Google Scholar] [CrossRef]
- Chung, H.S.; Kim, K.; Hong, S.S.; Hong, S.G.; Lee, K.; Chong, Y. The sul1 gene in Stenotrophomonas maltophilia with high-level resistance to trimethoprim/sulfamethoxazole. Ann. Lab. Med. 2015, 35, 246–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charoenlap, N.; Jiramonai, L.; Chittrakanwong, J.; Tunsakul, N.; Mongkolsuk, S.; Vattanaviboon, P. Inactivation of ahpC renders Stenotrophomonas maltophilia resistant to the disinfectant hydrogen peroxide. Antonie Van Leeuwenhoek 2019, 112, 809–814. [Google Scholar] [CrossRef] [PubMed]
- Cystic Fibrosis Foundation 2019 Patient Registry Annual Data Report. Available online: https://www.cff.org/About-Us/Reports-and-Financials/2019-Annual-Report/ (accessed on 27 April 2021).
- Hatziagorou, E.; Orenti, A.; Drevinek, P.; Kashirskaya, N.; Mei-Zahav, M.; De Boeck, K.; Pfleger, A.; Sciensano, M.T.; Lammertyn, E.; Macek, M.; et al. Changing epidemiology of the respiratory bacteriology of patients with cystic fibrosis–data from the European cystic fibrosis society patient registry. J. Cyst. Fibros. 2020, 19, 376–383. [Google Scholar] [CrossRef]
- Cystic Fibrosis Canada The Canadian Cystic Fibrosis Registry: 2019 Annual Data Report. Available online: https://www.cysticfibrosis.ca/our-programs/cf-registry (accessed on 27 April 2021).
- Hansen, C.R. Stenotrophomonas maltophilia: To be or not to be a cystic fibrosis pathogen. Curr. Opin. Pulm. Med. 2012, 18, 628–631. [Google Scholar] [CrossRef] [PubMed]
- Berdah, L.; Taytard, J.; Leyronnas, S.; Clement, A.; Boelle, P.Y.; Corvol, H. Stenotrophomonas maltophilia: A marker of lung disease severity. Pediatr. Pulmonol. 2018, 53, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Waters, V.; Atenafu, E.G.; Lu, A.; Yau, Y.; Tullis, E.; Ratjen, F. Chronic Stenotrophomonas maltophilia infection and mortality or lung transplantation in cystic fibrosis patients. J. Cyst. Fibros. 2013, 12, 482–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talmaciu, I.; Varlotta, L.; Mortensen, J.; Schidlow, D.V. Risk factors for emergence of Stenotrophomonas maltophilia in cystic fibrosis. Pediatr. Pulmonol. 2000, 30, 10–15. [Google Scholar] [CrossRef]
- Waters, V.J.; Gómez, M.I.; Soong, G.; Amin, S.; Ernst, R.K.; Prince, A. Immunostimulatory properties of the emerging pathogen Stenotrophomonas maltophilia. Infect. Immun. 2007, 75, 1698–1703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDaniel, M.S.; Schoeb, T.; Swords, W.E. Cooperativity between Stenotrophomonas maltophilia and Pseudomonas aeruginosa during polymicrobial airway infections. Infect. Immun. 2020, 88. [Google Scholar] [CrossRef]
- Kataoka, D.; Fujiwara, H.; Kawakami, T.; Tanaka, Y.; Tanimoto, A.; Ikawa, S.; Tanaka, Y. The indirect pathogenicity of Stenotrophomonas maltophilia. Int. J. Antimicrob. Agents 2003, 22, 601–606. [Google Scholar] [CrossRef] [Green Version]
- Yin, C.; Yang, W.; Meng, J.; Lv, Y.; Wang, J.; Huang, B. Co-infection of Pseudomonas aeruginosa and Stenotrophomonas maltophilia in hospitalised pneumonia patients has a synergic and significant impact on clinical outcomes. Eur. J. Clin. Microbiol. Infect. Dis. 2017, 36, 2231–2235. [Google Scholar] [CrossRef]
- Ryan, R.P.; Fouhy, Y.; Garcia, B.F.; Watt, S.A.; Niehaus, K.; Yang, L.; Tolker-Nielsen, T.; Dow, J.M. Interspecies signalling via the Stenotrophomonas maltophilia diffusible signal factor influences biofilm formation and polymyxin tolerance in Pseudomonas aeruginosa. Mol. Microbiol. 2008, 68, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Martínez, P.; Huedo, P.; Martinez-Servat, S.; Planell, R.; Ferrer-Navarro, M.; Daura, X.; Yero, D.; Gibert, I. Stenotrophomonas maltophilia responds to exogenous AHL signals through the LuxR solo SmoR (Smlt1839). Front. Cell. Infect. Microbiol. 2015, 5, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trifonova, A.; Strateva, T. Stenotrophomonas maltophilia—A low-grade pathogen with numerous virulence factors. Infect. Dis. 2019, 51, 168–178. [Google Scholar] [CrossRef] [PubMed]
- García, C.A.; Alcaraz, E.S.; Franco, M.A.; De Rossi, B.N.P. Iron is a signal for Stenotrophomonas maltophilia biofilm formation, oxidative stress response, OMPs expression, and virulence. Front. Microbiol. 2015, 6, 926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalidasan, V.; Joseph, N.; Kumar, S.; Awang Hamat, R.; Neela, V.K. Iron and virulence in Stenotrophomonas maltophilia: All we know so far. Front. Cell. Infect. Microbiol. 2018, 8, 401. [Google Scholar] [CrossRef] [PubMed]
- Crossman, L.C.; Gould, V.C.; Dow, J.M.; Vernikos, G.S.; Okazaki, A.; Sebaihia, M.; Saunders, D.; Arrowsmith, C.; Carver, T.; Peters, N.; et al. The complete genome, comparative and functional analysis of Stenotrophomonas maltophilia reveals an organism heavily shielded by drug resistance determinants. Genome Biol. 2008, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer-Navarro, M.; Planell, R.; Yero, D.; Mongiardini, E.; Torrent, G.; Huedo, P.; Martínez, P.; Roher, N.; Mackenzie, S.; Gibert, I.; et al. Abundance of the quorum-sensing factor Ax21 in four strains of Stenotrophomonas maltophilia correlates with mortality rate in a new zebrafish model of infection. PLoS ONE 2013, 8, 67207. [Google Scholar] [CrossRef] [Green Version]
- Winn, A.M.; Wilkinson, S.G. Structures of the O4 and O18 antigens of Stenotrophomonas maltophilia: A case of enantiomeric repeating units. Carbohydr. Res. 2001, 330, 215–221. [Google Scholar] [CrossRef]
- McKay, G.A.; Woods, D.E.; MacDonald, K.L.; Poole, K. Role of phosphoglucomutase of Stenotrophomonas maltophilia in lipopolysaccharide biosynthesis, virulence, and antibiotic resistance. Infect. Immun. 2003, 71, 3068–3075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, T.P.; Somers, E.B.; Wong, A.C.L. Differential biofilm formation and motility associated with lipopolysaccharide/exopolysaccharide-coupled biosynthetic genes in Stenotrophomonas maltophilia. J. Bacteriol. 2006, 188, 3116–3120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira-Garcia, D.; Dall’Agnol, M.; Rosales, M.; Azzuz, A.C.G.S.; Martinez, M.B.; Girón, J.A. Characterization of flagella produced by clinical strains of Stenotrophomonas maltophilia. Emerg. Infect. Dis. 2002, 8, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Zgair, A.K.; Chhibber, S. Adhesion of Stenotrophomonas maltophilia to mouse tracheal mucus is mediated through flagella. J. Med. Microbiol. 2011, 60, 1032–1037. [Google Scholar] [CrossRef]
- Pompilio, A.; Crocetta, V.; Confalone, P.; Nicoletti, M.; Petrucca, A.; Guarnieri, S.; Fiscarelli, E.; Savini, V.; Piccolomini, R.; Di Bonaventura, G. Adhesion to and biofilm formation on IB3-1 bronchial cells by Stenotrophomonas maltophilia isolates from cystic fibrosis patients. BMC Microbiol. 2010, 10. [Google Scholar] [CrossRef] [Green Version]
- De Oliveira-Garcia, D.; Dall’Agnol, M.; Rosales, M.; Azzuz, A.C.G.S.; Alcántara, N.; Martinez, M.B.; Girón, J.A. Fimbriae and adherence of Stenotrophomonas maltophilia to epithelial cells and to abiotic surfaces. Cell. Microbiol. 2003, 5, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Nicoletti, M.; Iacobino, A.; Prosseda, G.; Fiscarelli, E.; Zarrilli, R.; De Carolis, E.; Petrucca, A.; Nencioni, L.; Colonna, B.; Casalino, M. Stenotrophomonas maltophilia strains from cystic fibrosis patients: Genomic variability and molecular characterization of some virulence determinants. Int. J. Med. Microbiol. 2011, 301, 34–43. [Google Scholar] [CrossRef]
- Giltner, C.L.; Nguyen, Y.; Burrows, L.L. Type IV pilin proteins: Versatile molecular modules. Microbiol. Mol. Biol. Rev. 2012, 76, 740–772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalidasan, V.; Neela, V.K. Twitching motility of Stenotrophomonas maltophilia under iron limitation: In-silico, phenotypic and proteomic approaches. Virulence 2020, 11, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueirêdo, P.M.S.; Furumura, M.T.; Santos, A.M.; Sousa, A.C.T.; Kota, D.J.; Levy, C.E.; Yano, T. Cytotoxic activity of clinical Stenotrophomonas maltophilia. Lett. Appl. Microbiol. 2006, 43, 443–449. [Google Scholar] [CrossRef] [PubMed]
- Windhorst, S.; Frank, E.; Georgieva, D.N.; Genov, N.; Buck, F.; Borowski, P.; Weber, W. The major extracellular protease of the nosocomial pathogen Stenotrophomonas maltophilia: Characterization of the protein and molecular cloning of the gene. J. Biol. Chem. 2002, 277, 11042–11049. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuMont, A.L.; Karaba, S.M.; Cianciotto, N.P. Type II secretion-dependent degradative and cytotoxic activities mediated by Stenotrophomonas maltophilia serine proteases StmPr1 and StmPr2. Infect. Immun. 2015, 83, 3825–3837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DuMont, A.L.; Cianciotto, N.P. Stenotrophomonas maltophilia serine protease StmPr1 induces matrilysis, anoikis, and protease-activated receptor 2 activation in human lung epithelial cells. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaba, S.M.; White, R.C.; Cianciotto, N.P. Stenotrophomonas maltophilia encodes a type II protein secretion system that promotes detrimental effects on lung epithelial cells. Infect. Immun. 2013, 81, 3210–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nas, M.Y.; White, R.C.; DuMont, A.L.; Lopez, A.E.; Cianciottoa, N.P. Stenotrophomonas maltophilia encodes a VirB/VirD4 type IV secretion system that modulates apoptosis in human cells and promotes competition against heterologous bacteria, including Pseudomonas aeruginosa. Infect. Immun. 2019, 87. [Google Scholar] [CrossRef] [Green Version]
- Bayer-Santos, E.; Cenens, W.; Matsuyama, B.Y.; Oka, G.U.; Di Sessa, G.; Del Valle Mininel, I.; Alves, T.L.; Farah, C.S. The opportunistic pathogen Stenotrophomonas maltophilia utilizes a type IV secretion system for interbacterial killing. PLoS Pathog. 2019, 15. [Google Scholar] [CrossRef] [Green Version]
- Costa, T.R.D.; Felisberto-Rodrigues, C.; Meir, A.; Prevost, M.S.; Redzej, A.; Trokter, M.; Waksman, G. Secretion systems in Gram-negative bacteria: Structural and mechanistic insights. Nat. Rev. Microbiol. 2015, 13, 343–359. [Google Scholar] [CrossRef]
- Barber, C.E.; Tang, J.L.; Feng, J.X.; Pan, M.Q.; Wilson, T.J.G.; Slater, H.; Dow, J.M.; Williams, P.; Daniels, M.J. A novel regulatory system required for pathogenicity of Xanthomonas campestris is mediated by a small diffusible signal molecule. Mol. Microbiol. 1997, 24, 555–566. [Google Scholar] [CrossRef]
- An, S.Q.; Tang, J.L. Diffusible signal factor signaling regulates multiple functions in the opportunistic pathogen Stenotrophomonas maltophilia. BMC Res. Notes 2018, 11, 569. [Google Scholar] [CrossRef]
- Alcaraz, E.; García, C.; Friedman, L.; De Rossi, B.P. The rpf/DSF signalling system of Stenotrophomonas maltophilia positively regulates biofilm formation, production of virulence-associated factors and β-lactamase induction. FEMS Microbiol. Lett. 2019, 366, 69. [Google Scholar] [CrossRef]
- Yero, D.; Huedo, P.; Conchillo-Solé, O.; Martínez-Servat, S.; Mamat, U.; Coves, X.; Llanas, F.; Roca, I.; Vila, J.; Schaible, U.E.; et al. Genetic variants of the DSF quorum sensing system in Stenotrophomonas maltophilia influence virulence and resistance phenotypes among genotypically diverse clinical isolates. Front. Microbiol. 2020, 11, 1160. [Google Scholar] [CrossRef]
- Huedo, P.; Yero, D.; Martínez-Servat, S.; Estibariz, I.; Planell, R.; Martínez, P.; Ruyra, À.; Roher, N.; Roca, I.; Vila, J.; et al. Two different rpf clusters distributed among a population of Stenotrophomonas maltophilia clinical strains display differential diffusible signal factor production and virulence regulation. J. Bacteriol. 2014, 196, 2431–2442. [Google Scholar] [CrossRef] [Green Version]
- Huedo, P.; Coves, X.; Daura, X.; Gibert, I.; Yero, D. Quorum sensing signaling and quenching in the multidrug-resistant pathogen Stenotrophomonas maltophilia. Front. Cell. Infect. Microbiol. 2018, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Devos, S.; Van Oudenhove, L.; Stremersch, S.; Van Putte, W.; De Rycke, R.; Van Driessche, G.; Vitse, J.; Raemdonck, K.; Devreese, B. The effect of imipenem and diffusible signaling factors on the secretion of outer membrane vesicles and associated Ax21 proteins in Stenotrophomonas maltophilia. Front. Microbiol. 2015, 6, 298. [Google Scholar] [CrossRef] [Green Version]
- Ferrer-Navarro, M.; Torrent, G.; Mongiardini, E.; Conchillo-Solé, O.; Gibert, I.; Daura, X. Proteomic analysis of outer membrane proteins and vesicles of a clinical isolate and a collection strain of Stenotrophomonas maltophilia. J. Proteom. 2016, 142, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Jeon, H.; Na, S.H.; Kwon, H.I.; Selasi, G.N.; Nicholas, A.; Park, T.I.; Lee, S.H.; Lee, J.C. Stenotrophomonas maltophilia outer membrane vesicles elicit a potent inflammatory response in vitro and in vivo. Pathog. Dis. 2016, 74, 104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, S.-Q.; Tang, J. liang The Ax21 protein influences virulence and biofilm formation in Stenotrophomonas maltophilia. Arch. Microbiol. 2018, 200, 183–187. [Google Scholar] [CrossRef] [Green Version]
- Nikaido, H.; Vaara, M. Molecular basis of bacterial outer membrane permeability. Microbiol. Rev. 1985, 49, 1–32. [Google Scholar] [CrossRef]
- Sánchez, M.B. Antibiotic resistance in the opportunistic pathogen Stenotrophomonas maltophilia. Front. Microbiol. 2015, 6, 658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Z.; Zhang, L.; Poole, K. SmeC, an outer membrane multidrug efflux protein of Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2002, 46, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.; Martinez, J.L. Coning and characterization of SmeDEF, a novel multidrug efflux pump from Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2000, 44, 3079–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Li, X.; Poole, K. SmeDEF multidrug efflux pump contributes to intrinsic multidrug resistance in Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2001, 45, 3497–3503. [Google Scholar] [CrossRef] [Green Version]
- Blanco, P.; Corona, F.; Martínez, J.L. Involvement of the RND efflux pump transporter SmeH in the acquisition of resistance to ceftazidime in Stenotrophomonas maltophilia. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Gould, V.C.; Okazaki, A.; Avison, M.B. Coordinate hyperproduction of SmeZ and SmeJK efflux pumps extends drug resistance in Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2013, 57, 655–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, C.W.; Huang, Y.W.; Hu, R.M.; Yang, T.C. SmeOP-TolCSm efflux pump contributes to the multidrug resistance of Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2014, 58, 2405–2408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.H.; Huang, C.C.; Chung, T.C.; Hu, R.M.; Huang, Y.W.; Yang, T.C. Contribution of resistance-nodulation-division efflux pump operon smeU1-V-W-U2-X to multidrug resistance of Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2011, 55, 5826–5833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.T.; Huang, Y.W.; Chen, S.J.; Chang, C.W.; Yang, T.C. The SmeYZ efflux pump of Stenotrophomonas maltophilia contributes to drug resistance, virulence-related characteristics, and virulence in mice. Antimicrob. Agents Chemother. 2015, 59, 4067–4073. [Google Scholar] [CrossRef] [Green Version]
- Al-Hamad, A.; Upton, M.; Burnie, J. Molecular cloning and characterization of SmrA, a novel ABC multidrug efflux pump from Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 2009, 64, 731–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.T.; Huang, Y.W.; Liou, R.S.; Chang, Y.C.; Yang, T.C. MacABCsm, an ABC-type tripartite efflux pump of Stenotrophomonas maltophilia involved in drug resistance, oxidative and envelope stress tolerances and biofilm formation. J. Antimicrob. Chemother. 2014, 69, 3221–3226. [Google Scholar] [CrossRef]
- Huang, Y.W.; Hu, R.M.; Chu, F.Y.; Lin, H.R.; Yang, T.C. Characterization of a major facilitator superfamily (MFS) tripartite efflux pump EmrCABsm from Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 2013, 68, 2498–2505. [Google Scholar] [CrossRef] [Green Version]
- Hu, R.M.; Liao, S.T.; Huang, C.C.; Huang, Y.W.; Yang, T.C. An inducible fusaric acid tripartite efflux pump contributes to the fusaric acid resistance in Stenotrophomonas maltophilia. PLoS ONE 2012, 7. [Google Scholar] [CrossRef] [PubMed]
- Gil-Gil, T.; Martínez, J.L.; Blanco, P. Mechanisms of antimicrobial resistance in Stenotrophomonas maltophilia: A review of current knowledge. Expert Rev. Anti. Infect. Ther. 2020, 18, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, A.; Avison, M.B. Induction of L1 and L2 β-lactamase production in Stenotrophomonas maltophilia is dependent on an AmpR-type regulator. Antimicrob. Agents Chemother. 2008, 52, 1525–1528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avison, M.B.; Von Heldreich, C.J.; Higgins, C.S.; Bennett, P.M.; Walsh, T.R. A TEM-2 Beta-lactamase encoded on an active Tn1-like transposon in the genome of a clinical isolate of Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 2000, 46, 879–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Z.; Zhang, L.; McKay, G.A.; Poole, K. Role of the acetyltransferase AAC(6′)-Iz modifying enzyme in aminoglycoside resistance in Stenotrophomonas maltophilia. J. Antimicrob. Chemother. 2003, 51, 803–811. [Google Scholar] [CrossRef] [Green Version]
- Tada, T.; Miyoshi-Akiyama, T.; Dahal, R.K.; Mishra, S.K.; Shimada, K.; Ohara, H.; Kirikae, T.; Pokhrelc, B.M. Identification of a novel 6′-N-aminoglycoside acetyltransferase, AAC(6′)-Iak, from a multidrug-resistant clinical isolate of Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2014, 58, 6324–6327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okazaki, A.; Avison, M.B. Aph(3′)-IIc, an aminoglycoside resistance determinant from Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2007, 51, 359–360. [Google Scholar] [CrossRef] [Green Version]
- Valdezate, S.; Vindel, A.; Antonio Saéz-Nieto, J.; Baquero, F.; Cantó, R. Preservation of topoisomerase genetic sequences during in vivo and in vitro development of high-level resistance to ciprofloxacin in isogenic Stenotrophomonas maltophilia strains. J. Antimicrob. Chemother. 2005, 56, 220–223. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, K.; Kikuchi, K.; Sasaki, T.; Takahashi, N.; Ohtsuka, M.; Ono, Y.; Hiramatsu, K. Smqnr, a new chromosome-carried quinolone resistance gene in Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2008, 52, 3823–3825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sánchez, M.B.; Martínez, J.L. SmQnr contributes to intrinsic resistance to quinolones in Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 2010, 54, 580–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-León, G.; Salgado, F.; Oliveros, J.C.; Sánchez, M.B.; Martínez, J.L. Interplay between intrinsic and acquired resistance to quinolones in Stenotrophomonas maltophilia. Environ. Microbiol. 2014, 16, 1282–1296. [Google Scholar] [CrossRef] [PubMed]
- García-León, G.; Ruiz de Alegría Puig, C.; García de la Fuente, C.; Martínez-Martínez, L.; Martínez, J.L.; Sánchez, M.B. High-level quinolone resistance is associated with the overexpression of smeVWX in Stenotrophomonas maltophilia clinical isolates. Clin. Microbiol. Infect. 2015, 21, 464–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, L.F.; Chang, X.; Ye, Y.; Wang, Z.X.; Shao, Y.B.; Shi, W.; Li, X.; Li, J. Bin Stenotrophomonas maltophilia resistance to trimethoprim/sulfamethoxazole mediated by acquisition of sul and dfrA genes in a plasmid-mediated class 1 integron. Int. J. Antimicrob. Agents 2011, 37, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Barbolla, R.; Catalano, M.; Orman, B.E.; Famiglietti, A.; Vay, C.; Smayevsky, J.; Centrón, D.; Piñeiro, S.A. Class 1 integrons increase trimethoprim-sulfamethoxazole MICs against epidemiologically unrelated Stenotrophomonas maltophilia isolates. Antimicrob. Agents Chemother. 2004, 48, 666–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toleman, M.A.; Bennett, P.M.; Bennett, D.M.C.; Jones, R.N.; Walsh, T.R. Global emergence of trimethoprim/sulfamethoxazole resistance in Stenotrophomonas maltophilia mediated by acquisition of sul genes. Emerg. Infect. Dis. 2007, 13, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Giles, A.; Foushee, J.; Lantz, E.; Gumina, G. Sulfonamide allergies. Pharmacy 2019, 7, 132. [Google Scholar] [CrossRef] [Green Version]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [Green Version]
- Chanishvili, N. Phage Therapy-History from Twort and d’Herelle through Soviet Experience to Current Approaches. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 2012; Volume 83, pp. 3–40. ISBN 9780123944382. [Google Scholar]
- Summers, W.C. The strange history of phage therapy. Bacteriophage 2012, 2, 130–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burrowes, B.; Harper, D.R.; Anderson, J.; McConville, M.; Enright, M.C. Bacteriophage therapy: Potential uses in the control of antibiotic-resistant pathogens. Expert Rev. Anti. Infect. Ther. 2011, 9, 775–785. [Google Scholar] [CrossRef]
- Casjens, S. Prophages and bacterial genomics: What have we learned so far? Mol. Microbiol. 2003, 49, 277–300. [Google Scholar] [CrossRef] [PubMed]
- Touchon, M.; Bernheim, A.; Rocha, E.P.C. Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J. 2016, 10, 2744–2754. [Google Scholar] [CrossRef] [Green Version]
- Ramisetty, B.C.M.; Sudhakari, P.A. Bacterial “grounded” prophages: Hotspots for genetic renovation and innovation. Front. Genet. 2019, 10, 12. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patil, P.P.; Midha, S.; Kumar, S.; Patil, P.B. Genome sequence of type strains of genus Stenotrophomonas. Front. Microbiol 2016, 7, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, D.L.; McCutcheon, J.G.; Stothard, P.; Dennis, J.J. Novel Stenotrophomonas maltophilia temperate phage DLP4 is capable of lysogenic conversion. BMC Genomics 2019, 20, 300. [Google Scholar] [CrossRef] [PubMed]
- McCutcheon, J.G.; Lin, A.; Dennis, J.J. Isolation and characterization of the novel bacteriophage AXL3 against Stenotrophomonas maltophilia. Int. J. Mol. Sci. 2020, 21, 6338. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the evolution of bacterial pathogens: From genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [Green Version]
- Beres, S.B.; Sylva, G.L.; Barbian, K.D.; Lei, B.; Hoff, J.S.; Mammarella, N.D.; Liu, M.Y.; Smoot, J.C.; Porcella, S.F.; Parkins, L.D.; et al. Genome sequence of a serotype M3 strain of group A Streptococcus: Phage-encoded toxins, the high-virulence phenotype, and clone emergence. Proc. Natl. Acad. Sci. USA 2002, 99, 10078–10083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayashi, T.; Makino, K.; Ohnishi, M.; Kurokawa, K.; Ishii, K.; Yokoyama, K.; Han, C.G.; Ohtsubo, E.; Nakayama, K.; Murata, T.; et al. Complete genome sequence of enterohemorrhagic Escherichia coli O157:H7 and genomic comparison with a laboratory strain K-12. DNA Res. 2001, 8, 11–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, K.; Murase, K.; Sato, M.P.; Toyoda, A.; Itoh, T.; Mainil, J.G.; Piérard, D.; Yoshino, S.; Kimata, K.; Isobe, J.; et al. Differential dynamics and impacts of prophages and plasmids on the pangenome and virulence factor repertoires of Shiga toxin-producing Escherichia coli O145:H28. Microb. Genom. 2020, 6. [Google Scholar] [CrossRef] [PubMed]
- Waldor, M.K.; Mekalanos, J.J. Lysogenic conversion by a filamentous phage encoding cholera toxin. Science 1996, 272, 1910–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendrix, R.W.; Smith, M.C.M.; Burns, R.N.; Ford, M.E.; Hatfull, G.F. Evolutionary relationships among diverse bacteriophages and prophages: All the world’s a phage. Proc. Natl. Acad. Sci. USA 1999, 96, 2192–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ackermann, H.W.; Prangishvili, D. Prokaryote viruses studied by electron microscopy. Arch. Virol. 2012, 157, 1843–1849. [Google Scholar] [CrossRef]
- Roach, D.R.; Debarbieux, L. Phage therapy: Awakening a sleeping giant. Emerg. Top. Life Sci. 2017, 1, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yehl, K.; Lemire, S.; Yang, A.C.; Ando, H.; Mimee, M.; Torres, M.D.T.; de la Fuente-Nunez, C.; Lu, T.K. Engineering phage host-range and suppressing bacterial resistance through phage tail fiber mutagenesis. Cell 2019, 179, 459–469.e9. [Google Scholar] [CrossRef] [PubMed]
- Lenneman, B.R.; Fernbach, J.; Loessner, M.J.; Lu, T.K.; Kilcher, S. Enhancing phage therapy through synthetic biology and genome engineering. Curr. Opin. Biotechnol. 2021, 68, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Manrique, P.; Bolduc, B.; Walk, S.T.; Van Der Oost, J.; De Vos, W.M.; Young, M.J. Healthy human gut phageome. Proc. Natl. Acad. Sci. USA 2016, 113, 10400–10405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, R.A.; Vega, A.A.; Norman, H.M.; Ohaeri, M.; Levi, K.; Dinsdale, E.A.; Cinek, O.; Aziz, R.K.; McNair, K.; Barr, J.J.; et al. Global phylogeography and ancient evolution of the widespread human gut virus crAssphage. Nat. Microbiol. 2019, 4, 1727–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, S.; Baker, K.; Padman, B.S.; Patwa, R.; Dunstan, R.A.; Weston, T.A.; Schlosser, K.; Bailey, B.; Lithgow, T.; Lazarou, M.; et al. Bacteriophage transcytosis provides a mechanism to cross epithelial cell layers. MBio 2017, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luong, T.; Salabarria, A.C.; Roach, D.R. Phage therapy in the resistance era: Where do we stand and where are we going? Clin. Ther. 2020, 42, 1659–1680. [Google Scholar] [CrossRef]
- Aslam, S.; Lampley, E.; Wooten, D.; Karris, M.; Benson, C.; Strathdee, S.; Schooley, R.T. Lessons learned from the first 10 consecutive cases of intravenous bacteriophage therapy to treat multidrug-resistant bacterial infections at a single center in the United States. Open Forum Infect. Dis. 2020, 7. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Stanley, G.; Modak, M.; Koff, J.L.; Turner, P.E. Bacteriophage therapy for infections in CF. Pediatr. Pulmonol. 2021, 56, S4–S9. [Google Scholar] [CrossRef]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef] [Green Version]
- Loh, B.; Gondil, V.S.; Manohar, P.; Khan, F.M.; Yang, H.; Leptihn, S. Encapsulation and delivery of therapeutic phages. Appl. Environ. Microbiol. 2021, 87, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rosner, D.; Clark, J. Formulations for bacteriophage therapy and the potential uses of immobilization. Pharmaceuticals 2021, 14, 359. [Google Scholar] [CrossRef]
- Vinner, G.K.; Richards, K.; Leppanen, M.; Sagona, A.P.; Malik, D.J. Microencapsulation of enteric bacteriophages in a pH-responsive solid oral dosage formulation using a scalable membrane emulsification process. Pharmaceutics 2019, 11, 475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singla, S.; Harjai, K.; Katare, O.P.; Chhibber, S. Encapsulation of bacteriophage in liposome accentuates its entry in to macrophage and shields it from neutralizing antibodies. PLoS ONE 2016, 11, e0153777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chadha, P.; Katare, O.P.; Chhibber, S. Liposome loaded phage cocktail: Enhanced therapeutic potential in resolving Klebsiella pneumoniae mediated burn wound infections. Burns 2017, 43, 1532–1543. [Google Scholar] [CrossRef] [PubMed]
- Gordillo Altamirano, F.L.; Barr, J.J. Unlocking the next generation of phage therapy: The key is in the receptors. Curr. Opin. Biotechnol. 2021, 68, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shen, W.; Zhong, Q.; Chen, Q.; He, X.; Baker, J.L.; Xiong, K.; Jin, X.; Wang, J.; Hu, F.; et al. Development of a bacteriophage cocktail to constrain the emergence of phage-resistant Pseudomonas aeruginosa. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regeimbal, J.M.; Jacobs, A.C.; Corey, B.W.; Henry, M.S.; Thompson, M.G.; Pavlicek, R.L.; Quinones, J.; Hannah, R.M.; Ghebremedhin, M.; Crane, N.J.; et al. Personalized therapeutic cocktail of wild environmental phages rescues mice from Acinetobacter baumannii wound infections. Antimicrob. Agents Chemother. 2016, 60, 5806–5816. [Google Scholar] [CrossRef] [Green Version]
- Comeau, A.M.; Tétart, F.; Trojet, S.N.; Prère, M.F.; Krisch, H.M. Phage-antibiotic synergy (PAS): β-lactam and quinolone antibiotics stimulate virulent phage growth. PLoS ONE 2007, 2, e799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamal, F.; Dennis, J.J. Burkholderia cepacia complex phage-antibiotic synergy (PAS): Antibiotics stimulate lytic phage activity. Appl. Environ. Microbiol. 2015, 81, 1132–1138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, C.M.; McCutcheon, J.G.; Dennis, J.J. Aztreonam lysine increases the activity of phages E79 and phiKZ against Pseudomonas aeruginosa PA01. Microorganisms 2021, 9, 152. [Google Scholar] [CrossRef]
- Rasko, D.A.; Sperandio, V. Anti-virulence strategies to combat bacteria-mediated disease. Nat. Rev. 2010, 9, 117–128. [Google Scholar] [CrossRef]
- McCutcheon, J.G.; Peters, D.L.; Dennis, J.J. Identification and characterization of type IV pili as the cellular receptor of broad host range Stenotrophomonas maltophilia bacteriophages DLP1 and DLP2. Viruses 2018, 10, 338. [Google Scholar] [CrossRef] [Green Version]
- Gurney, J.; Brown, S.P.; Kaltz, O.; Hochberg, M.E. Steering phages to combat bacterial pathogens. Trends Microbiol. 2020, 28, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Gordillo Altamirano, F.; Forsyth, J.H.; Patwa, R.; Kostoulias, X.; Trim, M.; Subedi, D.; Archer, S.K.; Morris, F.C.; Oliveira, C.; Kielty, L.; et al. Bacteriophage-resistant Acinetobacter baumannii are resensitized to antimicrobials. Nat. Microbiol. 2021, 6, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Sistrom, M.; Wertz, J.E.; Kortright, K.E.; Narayan, D.; Turner, P.E. Phage selection restores antibiotic sensitivity in MDR Pseudomonas aeruginosa. Sci. Rep. 2016, 6, 26717. [Google Scholar] [CrossRef] [PubMed]
- Chan, B.K.; Turner, P.E.; Kim, S.; Mojibian, H.R.; Elefteriades, J.A.; Narayan, D. Phage treatment of an aortic graft infected with Pseudomonas aeruginosa. Evol. Med. Public Health 2018, 2018, 60–66. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, I.-Y.; Jang, H.-J.; Bae, H.-W.; Cho, Y.-H. A phage protein that inhibits the bacterial ATPase required for type IV pilus assembly. Proc. Natl. Acad. Sci. USA 2014, 111, 11503–11508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Smet, J.; Wagemans, J.; Hendrix, H.; Staes, I.; Visnapuu, A.; Horemans, B.; Aertsen, A.; Lavigne, R. Bacteriophage-mediated interference of the c-di-GMP signalling pathway in Pseudomonas aeruginosa. Microb. Biotechnol. 2021, 14, 967–978. [Google Scholar] [CrossRef]
- Moillo, A.M. Isolation of a transducing phage forming plaques on Pseudomonas maltophilia and Pseudomonas aeruginosa. Genet. Res. 1973, 21, 287–289. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Chen, C.R.; Lin, J.W.; Shen, G.H.; Chang, K.M.; Tseng, Y.H.; Weng, S.F. Isolation and characterization of novel giant Stenotrophomonas maltophilia phage ΦSMA5. Appl. Environ. Microbiol. 2005, 71, 1387–1393. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.R.; Lin, C.H.; Lin, J.W.; Chang, C.I.; Tseng, Y.H.; Weng, S.F. Characterization of a novel T4-type Stenotrophomonas maltophilia virulent phage Smp14. Arch. Microbiol. 2007, 188, 191–197. [Google Scholar] [CrossRef]
- Lee, C.-N.; Tseng, T.-T.; Chang, H.-C.; Lin, J.-W.; Weng, S.-F. Genomic sequence of temperate phage Smp131 of Stenotrophomonas maltophilia that has similar prophages in xanthomonads. BMC Microbiol. 2014, 14, 17. [Google Scholar] [CrossRef] [Green Version]
- García, P.; Monjardín, C.; Martín, R.; Madera, C.; Soberón, N.; Garcia, E.; Meana, Á.; Suárez, J.E. Isolation of new Stenotrophomonas bacteriophages and genomic characterization of temperate phage S1. Appl. Environ. Microbiol. 2008, 74, 7552–7560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.; Huang, Y.; Mi, Z.; Yin, X.; Wang, L.; Fan, H.; Zhang, Z.; An, X.; Chen, J.; Tong, Y. Complete genome sequence of IME13, a Stenotrophomonas maltophilia bacteriophage with large burst size and unique plaque polymorphism. J. Virol. 2012, 86, 11392–11393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Fan, H.; Pei, G.; Fan, H.; Zhang, Z.; An, X.; Mi, Z.; Shi, T.; Tong, Y. Complete genome sequence of IME15, the first T7-Like bacteriophage lytic to pan-antibiotic-resistant Stenotrophomonas maltophilia. J. Virol. 2012, 86, 13839–13840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Li, X. Biological characteristics of phage SM1 for Stenotrophomonas maltophilia and its effect in animal infection model. Zhejiang Da Xue Xue Bao Yi Xue Ban 2013, 42, 331–336. [Google Scholar] [PubMed]
- Peters, D.L.; Stothard, P.; Dennis, J.J. The isolation and characterization of Stenotrophomonas maltophilia T4-like bacteriophage DLP6. PLoS ONE 2017, 12, e0173341. [Google Scholar] [CrossRef] [PubMed]
- Pedramfar, A.; Maal, K.B.; Mirdamadian, S.H. Phage therapy of corrosion-producing bacterium Stenotrophomonas maltophilia using isolated lytic bacteriophages. Anti Corros. Methods Mater. 2017, 64, 607–612. [Google Scholar] [CrossRef]
- Peters, D.L.; Dennis, J.J. Complete genome sequence of temperate Stenotrophomonas maltophilia bacteriophage DLP5. Genome Announc. 2018, 6, e00073-18. [Google Scholar] [CrossRef] [Green Version]
- Peters, D.L.; McCutcheon, J.G.; Dennis, J.J. Characterization of novel broad-host-range bacteriophage DLP3 specific to Stenotrophomonas maltophilia as a potential therapeutic agent. Front. Microbiol. 2020, 11, 1358. [Google Scholar] [CrossRef] [PubMed]
- Marquez, A.; Newkirk, H.; Moreland, R.; Gonzalez, C.F.; Liu, M.; Ramsey, J. Complete genome sequence of Stenotrophomonas maltophilia podophage Ponderosa. Microbiol. Resour. Announc. 2019, 8, e01032-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayden, A.; Martinez, N.; Moreland, R.; Liu, M.; Gonzalez, C.F.; Gill, J.J.; Ramsey, J. Complete genome sequence of Stenotrophomonas phage Pokken. Microbiol. Resour. Announc. 2019, 8, e01095-19. [Google Scholar] [CrossRef] [Green Version]
- Vicary, A.; Newkirk, H.; Moreland, R.; Gonzalez, C.F.; Liu, M.; Ramsey, J.; Leavitt, J. Complete genome sequence of Stenotrophomonas maltophilia myophage Moby. Microbiol. Resour. Announc. 2020, 9, e01422-19. [Google Scholar] [CrossRef] [Green Version]
- Garza, K.D.; Newkirk, H.; Moreland, R.; Gonzalez, C.F.; Liu, M.; Ramsey, J.; Leavitt, J. Complete genome sequence of Stenotrophomonas phage Mendera. Microbiol. Resour. Announc. 2020, 9, e01411-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Zhang, R.; Hu, Y.; Liu, Y.; Wang, L.; An, X.; Song, L.; Shi, T.; Fan, H.; Tong, Y.; et al. Biological characteristics and genomic analysis of a Stenotrophomonas maltophilia phage vB_SmaS_BUCT548. Virus Genes 2021, 57, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Zhang, Y.; Jiang, Y.; Wu, H.; Sun, W.; Huang, Y.-P. Characterization and genomic analysis of ɸSHP3, a new transposable bacteriophage infecting Stenotrophomonas maltophilia. J. Virol. 2021, 95. [Google Scholar] [CrossRef] [PubMed]
- Hagemann, M.; Hasse, D.; Berg, G. Detection of a phage genome carrying a zonula occludens like toxin gene (zot) in clinical isolates of Stenotrophomonas maltophilia. Arch. Microbiol. 2006, 185, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Q.; Shen, P.; Huang, Y.P. Isolation and characterization of a novel filamentous phage from Stenotrophomonas maltophilia. Arch. Virol. 2012, 157, 1643–1650. [Google Scholar] [CrossRef]
- Liu, J.; Chen, P.; Zheng, C.; Huang, Y.P. Characterization of maltocin P28, a novel phage tail-like bacteriocin from Stenotrophomonas maltophilia. Appl. Environ. Microbiol. 2013, 79, 5593–5600. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova, M.; Shcherbatova, N.; Kurakov, A.; Mindlin, S. Genomic characterization and integrative properties of phiSMA6 and phiSMA7, two novel filamentous bacteriophages of Stenotrophomonas maltophilia. Arch. Virol. 2014, 159, 1293–1303. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Krupovic, M.; Daly, R.A.; Borges, A.L.; Nayfach, S.; Schulz, F.; Sharrar, A.; Matheus Carnevali, P.B.; Cheng, J.F.; Ivanova, N.N.; et al. Cryptic inoviruses revealed as pervasive in bacteria and archaea across Earth’s biomes. Nat. Microbiol. 2019, 4, 1895–1906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgener, E.B.; Sweere, J.M.; Bach, M.S.; Secor, P.R.; Haddock, N.; Jennings, L.K.; Marvig, R.L.; Johansen, H.K.; Rossi, E.; Cao, X.; et al. Filamentous bacteriophages are associated with chronic Pseudomonas lung infections and antibiotic resistance in cystic fibrosis. Sci. Transl. Med. 2019, 11, 9748. [Google Scholar] [CrossRef] [PubMed]
- Sweere, J.M.; Van Belleghem, J.D.; Ishak, H.; Bach, M.S.; Popescu, M.; Sunkari, V.; Kaber, G.; Manasherob, R.; Suh, G.A.; Cao, X.; et al. Bacteriophage trigger antiviral immunity and prevent clearance of bacterial infection. Science 2019, 363. [Google Scholar] [CrossRef]
- Secor, P.R.; Burgener, E.B.; Kinnersley, M.; Jennings, L.K.; Roman-Cruz, V.; Popescu, M.; Van Belleghem, J.D.; Haddock, N.; Copeland, C.; Michaels, L.A.; et al. Pf bacteriophage and their impact on Pseudomonas virulence, mammalian immunity, and chronic infections. Front. Immunol. 2020, 11, 244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef]
- Lee, C.N.; Lin, J.W.; Chow, T.Y.; Tseng, Y.H.; Weng, S.F. A novel lysozyme from Xanthomonas oryzae phage φXo411 active against Xanthomonas and Stenotrophomonas. Protein Expr. Purif. 2006, 50, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Dams, D.; Brøndsted, L.; Drulis-Kawa, Z.; Briers, Y. Engineering of receptor-binding proteins in bacteriophages and phage tail-like bacteriocins. Biochem. Soc. Trans. 2019, 47, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, Y.; Yin, M.; Xu, Y.; Liang, X.; Huang, Y.P. Characterization of maltocin S16, a phage tail-like bacteriocin with antibacterial activity against Stenotrophomonas maltophilia and Escherichia coli. J. Appl. Microbiol. 2019, 127, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef] [PubMed]
- Burrows, L.L. Pseudomonas aeruginosa twitching motility: Type IV pili in action. Annu. Rev. Microbiol. 2012, 66, 493–520. [Google Scholar] [CrossRef] [Green Version]
- Comolli, J.C.; Hauser, A.R.; Waite, L.; Whitchurch, C.B.; Mattick, J.S.; Engel, J.N. Pseudomonas aeruginosa gene products PilT and PilU are required for cytotoxicity in vitro and virulence in a mouse model of acute pneumonia. Infect. Immun. 1999, 67, 3625–3630. [Google Scholar] [CrossRef] [Green Version]
- Mattick, J.S. Type IV pili and twitching motility. Annu. Rev. Microbiol. 2002, 56, 289–314. [Google Scholar] [CrossRef]
- Monteiro, R.; Pires, D.P.; Costa, A.R.; Azeredo, J. Phage therapy: Going temperate? Trends Microbiol. 2019, 27, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Mageeney, C.M.; Sinha, A.; Mosesso, R.A.; Medlin, D.L.; Lau, B.Y.; Rokes, A.B.; Lane, T.W.; Branda, S.S.; Williams, K.P. Computational basis for on-demand production of diversified therapeutic phage cocktails. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Pires, D.P.; Cleto, S.; Sillankorva, S.; Azeredo, J.; Lu, T.K. Genetically engineered phages: A review of advances over the last decade. Microbiol. Mol. Biol. Rev. 2016, 80, 523–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedrick, R.M.; Guerrero-Bustamante, C.A.; Garlena, R.A.; Russell, D.A.; Ford, K.; Harris, K.; Gilmour, K.C.; Soothill, J.; Jacobs-Sera, D.; Schooley, R.T.; et al. Engineered bacteriophages for treatment of a patient with a disseminated drug-resistant Mycobacterium abscessus. Nat. Med. 2019, 25, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.P.; Monteiro, R.; Mil-Homens, D.; Fialho, A.; Lu, T.K.; Azeredo, J. Designing P. aeruginosa synthetic phages with reduced genomes. Sci. Rep. 2021, 11, 1–10. [Google Scholar] [CrossRef]
- Liu, J.; Dehbi, M.; Moeck, G.; Arhin, F.; Banda, P.; Bergeron, D.; Callejo, M.; Ferretti, V.; Ha, N.; Kwan, T.; et al. Antimicrobial drug discovery through bacteriophage genomics. Nat. Biotechnol. 2004, 22, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Wan, X.; Hendrix, H.; Skurnik, M.; Lavigne, R. Phage-based target discovery and its exploitation towards novel antibacterial molecules. Curr. Opin. Biotechnol. 2021, 68, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Martel, B.; Moineau, S. CRISPR-Cas: An efficient tool for genome engineering of virulent bacteriophages. Nucleic Acids Res. 2014, 42, 9504–9513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemay, M.L.; Tremblay, D.M.; Moineau, S. Genome engineering of virulent lactococcal phages using CRISPR-Cas9. ACS Synth. Biol. 2017, 6, 1351–1358. [Google Scholar] [CrossRef]
- Burrowes, B.H.; Molineux, I.J.; Fralick, J.A. Directed in vitro evolution of therapeutic bacteriophages: The Appelmans protocol. Viruses 2019, 11, 241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Favor, A.H.; Llanos, C.D.; Youngblut, M.D.; Bardales, J.A. Optimizing bacteriophage engineering through an accelerated evolution platform. Sci. Rep. 2020, 10. [Google Scholar] [CrossRef] [PubMed]
- Seed, K.D.; Dennis, J.J. Experimental bacteriophage therapy increases survival of Galleria mellonella larvae infected with clinically relevant strains of the Burkholderia cepacia complex. Antimicrob. Agents Chemother. 2009, 53, 2205–2208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plackett, B. No money for new drugs. Nat. Outlook 2020, 586, S50–S52. [Google Scholar]
- McKenna, M. The Antibiotic Gamble. Nature 2020, 584, 338–341. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phage | Source; Isolation Strain | Genome Length (bp) | GC (%) | Family | Phage Relatedness 1 | Lifestyle | Unique Features | Reference; Accession If Applicable |
|---|---|---|---|---|---|---|---|---|
| M6 | P. maltophiliaa 6 | – | – | Siphoviridae | – | Temperate | First phage isolated for S. maltophilia Transducing phage Host range mutant, M6a, is capable of infecting P. aeruginosa | [151] |
| ΦSMA5 | Sputum; S. maltophilia T39 | ~250 kb b | – | Myoviridae | – | Virulent | Broad host range, 61 out of 87 strains susceptible Burst size 95 phages/cell DNA is restriction enzyme resistant | [152] |
| Smp14 | Sewage; S. maltophilia T14 | ~160 kb c | 53.3 c | Myoviridae | Stenotrophomonas phage YB07 | Virulent | T4-like phage Moderate host range infecting 37 out of 87 clinical isolates Adsorbs to poles of cells Burst size ~150 phages/cell DNA is restriction enzyme resistant | [153] DQ364602 |
| S1 | Environmental S. maltophilia CECT 4793 | 40,287 | 63.7 | Siphoviridae | <1% coverage to Stenotrophomonas phage Smp131 | Temperate | Narrow tropism, infecting 4 out of 26 strains Encodes putative GspM protein involved in host type II secretion system Burst size of ~75 phages/cell 48 ORFs | [155] NC_011589 |
| S3 | Sewage; S. maltophilia E539 | ~33 kb b | – | Myoviridae | – | Virulent | Moderate host range infecting 12 out of 26 strains Burst size ~100 phages/cell Short eclipse period of 30 min DNA is restriction enzyme resistant | [155] |
| S4 | Sewage; S. maltophilia F227 | ~200 kb b | – | Siphoviridae | – | Temperate | Broad host range infecting 18 out of 26 strains Burst size ~80 phages/cell DNA is restriction enzyme resistant | [155] |
| IME13 | Sewage; clinical S. maltophilia | 162,327 | 41.2 | Myoviridaed | >97% Aeromonas phage phiAS4 | Virulent | Large burst size >3000 phages/cell Plaque polymorphism with three plaque sizes 182 ORFs; 15 tRNAs | [156] JX306041 |
| IME15 | Sewage; clinical S. maltophilia | 38,513 | 53.7 | Podoviridaed | >97% Aeromonas phage PZL-Ah1 | Virulent | T7-like phage Burst size >100 phages/cell 45 ORFs | [157] JX872508 |
| SM1 | Sewage; S. maltophilia | ~50 kb b | – | Myoviridae | – | – | Large burst size of 187 phages/cell In vivo mouse trials show 100% of SM1 treated mice surviving past day 7 | [158] |
| Smp131 | Clinical S. maltophilia T13 | 33,525 | 65.0 | Myoviridae | Uncultured Caudovirales phage clone 3S_12 | Temperate | P2-like phage Narrow tropism, infecting 3 out of 86 strains 47 ORFs | [154] JQ809663 |
| DLP1 | Red Deer River sediment; clinical S. maltophilia D1585 | 42,887 | 53.7 | Siphoviridae | >97% to P. aeruginosa phage SCUT-S4 | Virulent | Host range crosses taxonomic orders to P. aeruginosa strains. Uses type IV pili as host receptor 57 ORFs | [11,144] KR537872 |
| DLP2 | Blue flax soil; clinical S. maltophilia D1585 | 42,593 | 53.7 | Siphoviridae | >97% to P. aeruginosa phage PA73 | Virulent | Host range crosses taxonomic orders to P. aeruginosa strains. Uses type IV pili as host receptor 58 ORFs | [11,144] KR537871 |
| DLP3 | Empty soil; clinical S. maltophilia D1571 | 96,852 | 58.3 | Siphoviridae | Stenotrophomonas phage DLP5 | Temperate | Uses type IV pili as host receptor Second member of the Delepquintavirus genus Broad host range infecting 22 out of 29 strains Therapeutically active in D1571 infected G. mellonella larvae Causes lysogenic conversion of D1571 Encodes functional erythromycin resistance protein DNA is restriction enzyme resistant 148 ORFs; 5 tRNAs | [162] MT110073 |
| DLP4 | Planter soil; clinical S. maltophilia D1585 | 63,945 | 65.1 | Siphoviridae | Xanthomonas phage Bosa | Temperate | Moderate host range infecting 14 out of 27 strains Uses type IV pili as host receptor Causes lysogenic conversion of host Encodes functional trimethoprim resistance protein and virulence factor YbiA DNA is restriction enzyme resistant 82 ORFs; 1 tRNA | [112] MG018224 |
| DLP5 | Empty soil; clinical S. maltophilia D1614 | 96,542 | 58.4 | Siphoviridae | Stenotrophomonas phage DLP3 | Temperate | Type strain of Delepquintavirus genus Temperate phage is maintained as a phagemid Narrow host range infecting 5 out of 27 strains Causes lysogenic conversion of D1614 Encodes putative erythromycin resistance protein DNA is restriction enzyme resistant 149 ORFs; 5 tRNAs | [161] NC_042082 |
| DLP6 | Planter soil; clinical S. maltophilia D1571 | 168,489 | 55.8 | Myoviridae | Sinorhizobium phage phiN3 | Virulent | Moderate host range infecting 13 out of 27 strains Divergent T4-like virus Encodes a transposase DNA is restriction enzyme resistant 241 ORFs; 30 tRNAs. | [159] KU682439 |
| AXL3 | Empty soil; clinical S. maltophilia D1585 | 47,545 | 63.3 | Siphoviridae | 4% coverage to Pseudomonas phage JG012 | Virulent | Narrow host range infecting 5 out of 29 strains Uses type IV pili as host receptor Long infection cycle with burst size of 38 phages/cell DNA is restriction enzyme resistant 65 ORFs | [113] MT536174 |
| Ponderosa | Water sample; S. maltophilia ATCC 17807 | 42,612 | 60.0 | Podoviridae | Xylella phage Paz | – | T7-like phage 54 ORFs | [163] MK903280 |
| Pokken | Water sample; S. maltophilia ATCC 17807 | 76,239 | 55.1 | Podoviridae | Xanthomonas phage RiverRider | – | 92 ORFs; 5 tRNAs | [164] MN062186 |
| Moby | Wastewater; S. maltophilia ATCC 17807 | 159,365 | 54.1 | Myoviridae | Stenotrophomonas phage Mendera | – | T4-like phage 271 ORFs; 24 tRNAs | [165] MN095772 |
| Mendera | Wastewater; S. maltophilia ATCC 17807 | 159,961 | 54.0 | Myoviridae | >97% to Stenotrophomonas phage YB07 | – | T4-like phage 287 ORFs; 23 tRNAs | [166] MN098328 |
| BUCT548 | S. maltophilia 824 | 62,354 | 56.3 | Siphoviridae | Stenotrophomonas phage Salva | – | Broad host range infecting 11 out of 13 strains Burst size 134 phages/cell 102 ORFs; 1 tRNA. | [167] MN937349 |
| phiSHP3 | S. maltophilia c31 | 37,611 | 65.3 | Siphoviridae | Pseudomonas phage B3 | Temperate | Transposable phage Moderate host range infecting 20 out of 83 strains 51 ORFs | [168] MT872956 |
| IME-SM1 | Hospital sewage | 159,514 | 54.1 | Ackermannviridae | >98% to Stenotrophomonas phage YB07 | - | 254 ORFs; 20 tRNAs. | Accession: KR560069 |
| YB07 | – | 159,862 | 54.1 | Ackermannviridae | >98% to Stenotrophomonas phage IME-SM1 | – | 257 ORFs | Accession: MK580972 |
| BUCT555 | Hospital sewage; S. maltophilia 1207 | 39,440 | 61.4 | Podoviridae | 2% coverage to Myxococcus phage Mx8 | – | 57 ORFs | Accession: MW291508 |
| Salva | Soil; S. maltophilia | 60,789 | 56.4 | Siphoviridae | Stenotrophomonas phage BUCT548 | – | 102 ORFs; 1 tRNA. | Accession: MW393850 |
| Filamentous phages | ||||||||
| ΦSMA9 | Clinical S. maltophilia c5 | 6907 | 62.4 | Inoviridae | Inoviridae sp. Isolate ctda6 | Chronic | Encodes zot-like protein 7 ORFs | [169] NC_007189 |
| ΦSHP1 | Environmental S. maltophilia P2 | 6867 | 61.1 | Inoviridae | Stenotrophomonas phage ΦSMA7 | Chronic | Encodes zot-like protein 10 ORFs | [170] NC_010429 |
| ΦSHP2 | - | 5819 | 61.5 | Inoviridae | Inoviridae sp. Isolate ctda6 | Chronic | Encodes zot-like protein 9 ORFs | [171] NC_015586 |
| ΦSMA6 | Environmental S. maltophilia Khak84 | 7648 | 62.6 | Inoviridae | Phage ΦSMA9 | Chronic | Encodes zot-like protein and putative conjugal transfer protein 11 ORFs | [172] HG315669 |
| ΦSMA7 | Environmental S. maltophilia Khak84 | 7069 | 62.3 | Inoviridae | Stenotrophomonas phage ΦSHP2 | Chronic | Encodes zot-like protein 11 ORFs | [172] HG007973 |
| Phage-derived antimicrobials and PTLBs | ||||||||
| Lys411 lysozyme | Xanthomonas oryzae phage ΦXo411 | 537 | 54.2 | – | X. oryzae phage Xp10 lysozyme | – | No holin required for export to periplasm 124,400 U/mg activity against S. maltophilia | [178] DQ408365 |
| Maltocin P28 | S. maltophilia P28 | 19,919 | 66.2 | – | – | – | Bactericidal activity against 38 out of 81 strains R-type pyocin structure Mitomycin C inducible, thermolabile, sensitive to proteinase K 23 ORFs | [171] KC787694 |
| Maltocin S16 | S. maltophilia S16 | 19,658 | 66.3 | – | – | – | Bactericidal activity against 62 out of 86 strains of S. maltophilia Also active against 8 out of 14 E. coli strains Mitomycin C inducible, thermolabile, insensitive to proteases Binds LPS as surface receptor 23 ORFs | [180] MH703584 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCutcheon, J.G.; Dennis, J.J. The Potential of Phage Therapy against the Emerging Opportunistic Pathogen Stenotrophomonas maltophilia. Viruses 2021, 13, 1057. https://doi.org/10.3390/v13061057
McCutcheon JG, Dennis JJ. The Potential of Phage Therapy against the Emerging Opportunistic Pathogen Stenotrophomonas maltophilia. Viruses. 2021; 13(6):1057. https://doi.org/10.3390/v13061057
Chicago/Turabian StyleMcCutcheon, Jaclyn G., and Jonathan J. Dennis. 2021. "The Potential of Phage Therapy against the Emerging Opportunistic Pathogen Stenotrophomonas maltophilia" Viruses 13, no. 6: 1057. https://doi.org/10.3390/v13061057
APA StyleMcCutcheon, J. G., & Dennis, J. J. (2021). The Potential of Phage Therapy against the Emerging Opportunistic Pathogen Stenotrophomonas maltophilia. Viruses, 13(6), 1057. https://doi.org/10.3390/v13061057