
Membrane-Associated Flavivirus Replication Complex—Its Organization and Regulation


{kind=link}
{kind=link}
Abstract
:1. Introduction
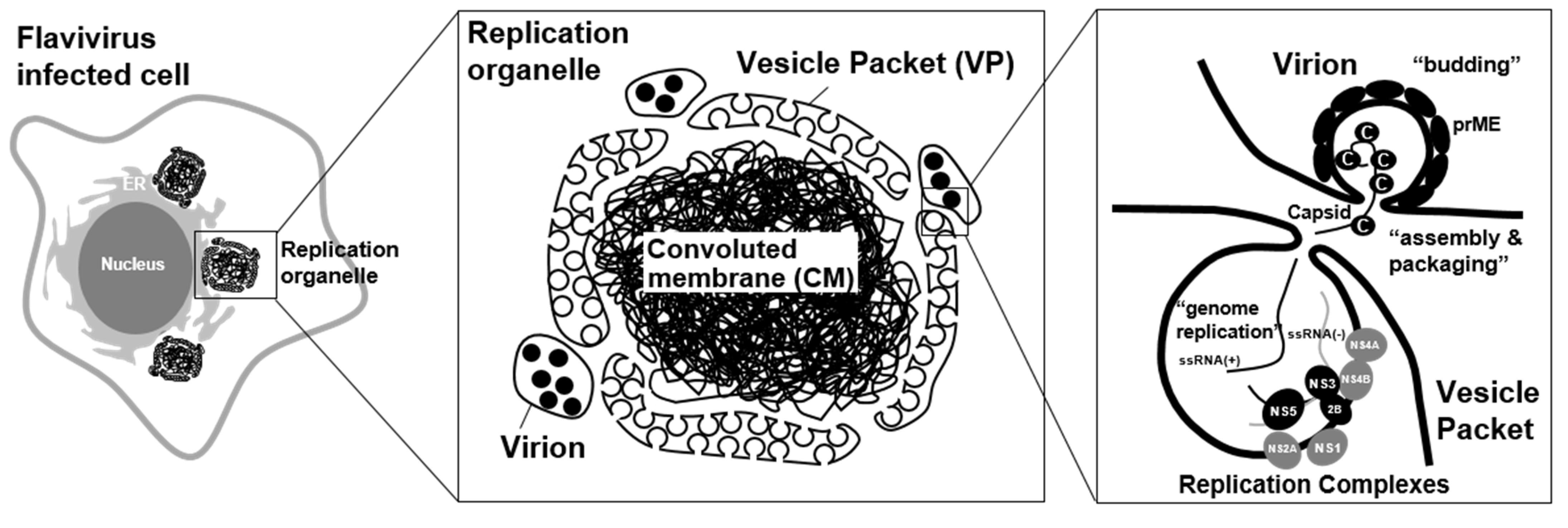
2. Flavivirus Infection Rearranges Intracellular Membrane System and Creates Organelle-Like Structures

3. Viral Proteins Involved in the Formation of a Viral Replication Organelle
4. Host ER Shaping Proteins Are Involved in the Formation of a Viral Replication Organelle
5. The Endosomal Sorting Complex Required for Transport (ESCRT) Proteins Is Potentially Involved in Membrane Invagination and Fission
6. Cellular Autophagic Machinery Is Activated in the Flavivirus-Infected Cells
7. Flavivirus Infection Changes the Intracellular Membrane Lipid Composition
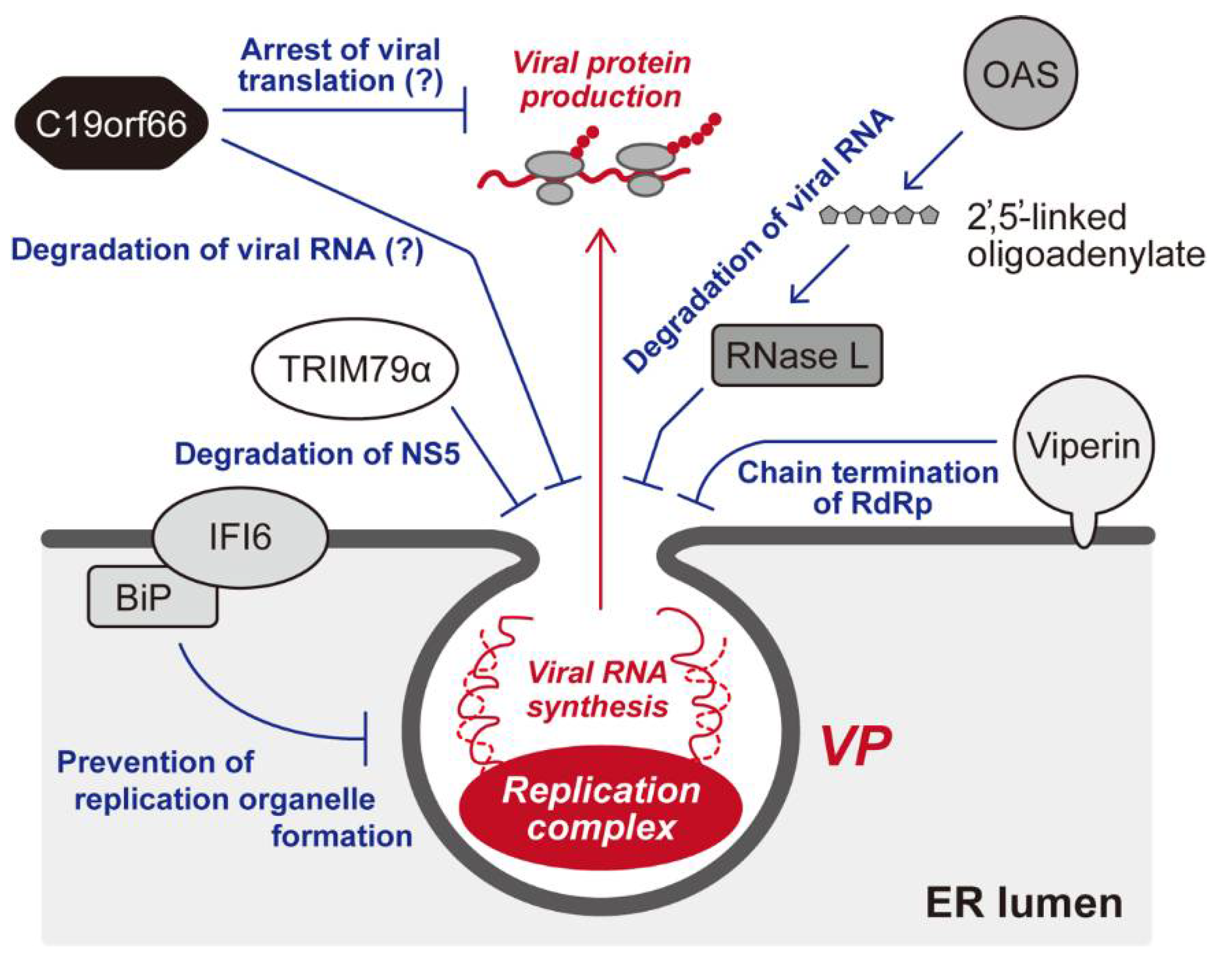
8. Cellular Restriction of the Membrane-Associated Flaviviral Replication Complex
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Neufeldt, C.J.; Cortese, M.; Acosta, E.G.; Bartenschlager, R. Rewiring cellular networks by members of the Flaviviridae family. Nat. Rev. Genet. 2018, 16, 125–142. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.A.; Jamieson, D.; Honein, M.A.; Petersen, L.R. Zika Virus and Birth Defects—Reviewing the Evidence for Causality. New Engl. J. Med. 2016, 374, 1981–1987. [Google Scholar] [CrossRef]
- García-Sastre, A. Ten Strategies of Interferon Evasion by Viruses. Cell Host Microbe 2017, 22, 176–184. [Google Scholar] [CrossRef]
- Perera-Lecoin, M.; Meertens, L.; Carnec, X.; Amara, A. Flavivirus Entry Receptors: An Update. Viruses 2013, 6, 69–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollati, M.; Alvarez, K.; Assenberg, R.; Baronti, C.; Canard, B.; Cook, S.; Coutard, B.; Decroly, E.; de Lamballerie, X.; Gould, E.A.; et al. Structure and functionality in flavivirus NS-proteins: Perspectives for drug design. Antivir. Res. 2010, 87, 125–148. [Google Scholar] [CrossRef]
- Murray, C.L.; Jones, C.T.; Rice, C.M. Architects of assembly: Roles of Flaviviridae non-structural proteins in virion morphogenesis. Nat. Rev. Genet. 2008, 6, 699–708. [Google Scholar] [CrossRef] [Green Version]
- Egloff, M.-P.; Decroly, E.; Malet, H.; Selisko, B.; Benarroch, D.; Ferron, F.; Canard, B. Structural and Functional Analysis of Methylation and 5′-RNA Sequence Requirements of Short Capped RNAs by the Methyltransferase Domain of Dengue Virus NS5. J. Mol. Biol. 2007, 372, 723–736. [Google Scholar] [CrossRef]
- Yu, I.-M.; Zhang, W.; Holdaway, H.A.; Li, L.; Kostyuchenko, V.A.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G.; Chen, J. Structure of the Immature Dengue Virus at Low pH Primes Proteolytic Maturation. Science 2008, 319, 1834–1837. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Lok, S.-M.; Yu, I.-M.; Zhang, Y.; Kuhn, R.J.; Chen, J.; Rossmann, M.G. The Flavivirus Precursor Membrane-Envelope Protein Complex: Structure and Maturation. Science 2008, 319, 1830–1834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aktepe, T.E.; MacKenzie, J.M. Shaping the flavivirus replication complex: It is curvaceous! Cell. Microbiol. 2018, 20, e12884. [Google Scholar] [CrossRef] [Green Version]
- Yi, Z.; Sperzel, L.; Nürnberger, C.; Bredenbeek, P.J.; Lubick, K.J.; Best, S.M.; Stoyanov, C.T.; Law, L.M.J.; Yuan, Z.; Rice, C.M.; et al. Identification and Characterization of the Host Protein DNAJC14 as a Broadly Active Flavivirus Replication Modulator. PLoS Pathog. 2011, 7, e1001255. [Google Scholar] [CrossRef] [Green Version]
- Rothan, H.A.; Kumar, M. Role of Endoplasmic Reticulum-Associated Proteins in Flavivirus Replication and Assembly Complexes. Pathogens 2019, 8, 148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackenzie, J.M.; Khromykh, A.A.; Jones, M.; Westaway, E.G. Subcellular Localization and Some Biochemical Properties of the Flavivirus Kunjin Nonstructural Proteins NS2A and NS4A. Virology 1998, 245, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and Three-Dimensional Architecture of the Dengue Virus Replication and Assembly Sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef] [Green Version]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; MacKenzie, J.M. The Endoplasmic Reticulum Provides the Membrane Platform for Biogenesis of the Flavivirus Replication Complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef] [Green Version]
- MacKenzie, J. Wrapping Things up about Virus RNA Replication. Traffic 2005, 6, 967–977. [Google Scholar] [CrossRef]
- Miller, S.; Locker, J.K. Modification of intracellular membrane structures for virus replication. Nat. Rev. Microbiol. 2008, 6, 363–374. [Google Scholar] [CrossRef]
- Salonen, A.; Ahola, T.; Kääriäinen, L. Viral RNA Replication in Association with Cellular Membranes. Curr. Top. Microbiol. Immunol. 2004, 285, 139–173. [Google Scholar] [CrossRef]
- Boon, J.A.D.; Diaz, A.; Ahlquist, P. Cytoplasmic Viral Replication Complexes. Cell Host Microbe 2010, 8, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Nazmi, A.; Dutta, K.; Basu, A. RIG-I Mediates Innate Immune Response in Mouse Neurons Following Japanese Encephalitis Virus Infection. PLoS ONE 2011, 6, e21761. [Google Scholar] [CrossRef] [Green Version]
- Netherton, C.; Moffat, K.; Brooks, E.; Wileman, T. A Guide to Viral Inclusions, Membrane Rearrangements, Factories, and Viroplasm Produced During Virus Replication. Int. Rev. Cytol. 2007, 70, 101–182. [Google Scholar] [CrossRef]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [Green Version]
- Överby, A.K.; Popov, V.L.; Niedrig, M.; Weber, F. Tick-Borne Encephalitis Virus Delays Interferon Induction and Hides Its Double-Stranded RNA in Intracellular Membrane Vesicles. J. Virol. 2010, 84, 8470–8483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoenen, A.; Liu, W.; Kochs, G.; Khromykh, A.A.; MacKenzie, J.M. West Nile virus-induced cytoplasmic membrane structures provide partial protection against the interferon-induced antiviral MxA protein. J. Gen. Virol. 2007, 88, 3013–3017. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef] [Green Version]
- Arakawa, M.; Morita, E. Flavivirus Replication Organelle Biogenesis in the Endoplasmic Reticulum: Comparison with Other Single-Stranded Positive-Sense RNA Viruses. Int. J. Mol. Sci. 2019, 20, 2336. [Google Scholar] [CrossRef] [Green Version]
- Romero-Brey, I.; Bartenschlager, R. Membranous Replication Factories Induced by Plus-Strand RNA Viruses. Viruses 2014, 6, 2826–2857. [Google Scholar] [CrossRef]
- Chatel-Chaix, L.; Bartenschlager, R. Dengue Virus- and Hepatitis C Virus-Induced Replication and Assembly Compartments: The Enemy Inside—Caught in the Web. J. Virol. 2014, 88, 5907–5911. [Google Scholar] [CrossRef] [Green Version]
- Junjhon, J.; Pennington, J.G.; Edwards, T.J.; Perera, R.; Lanman, J.; Kuhn, R.J. Ultrastructural Characterization and Three-Dimensional Architecture of Replication Sites in Dengue Virus-Infected Mosquito Cells. J. Virol. 2014, 88, 4687–4697. [Google Scholar] [CrossRef] [Green Version]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural characterization of Zika virus replication factories. Cell Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef] [Green Version]
- Offerdahl, D.K.; Dorward, D.W.; Hansen, B.T.; Bloom, M.E. A Three-Dimensional Comparison of Tick-Borne Flavivirus Infection in Mammalian and Tick Cell Lines. PLoS ONE 2012, 7, e47912. [Google Scholar] [CrossRef]
- Płaszczyca, A.; Scaturro, P.; Neufeldt, C.J.; Cortese, M.; Cerikan, B.; Ferla, S.; Brancale, A.; Pichlmair, A.; Bartenschlager, R. A novel interaction between dengue virus nonstructural protein 1 and the NS4A-2K-4B precursor is required for viral RNA replication but not for formation of the membranous replication organelle. PLoS Pathog. 2019, 15, e1007736. [Google Scholar] [CrossRef]
- Stern, O.; Hung, Y.-F.; Valdau, O.; Yaffe, Y.; Harris, E.; Hoffmann, S.; Willbold, D.; Sklan, E.H. An N-Terminal Amphipathic Helix in Dengue Virus Nonstructural Protein 4A Mediates Oligomerization and Is Essential for Replication. J. Virol. 2013, 87, 4080–4085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melo, C.; De Oliveira, D.N.; Lima, E.D.O.; Guerreiro, T.; Esteves, C.Z.; Beck, R.M.; Padilla, M.A.; Milanez, G.; Arns, C.W.; Proenca-Modena, J.L.; et al. A Lipidomics Approach in the Characterization of Zika-Infected Mosquito Cells: Potential Targets for Breaking the Transmission Cycle. PLoS ONE 2016, 11, e0164377. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, Z.; Chukkapalli, V.; Nchoutmboube, J.; Li, J.; Randall, G.; Belov, G.A.; Wang, X. Positive-strand RNA viruses stimulate host phosphatidylcholine synthesis at viral replication sites. Proc. Natl. Acad. Sci. USA 2016, 113, E1064–E1073. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-D.; Ye, H.-Q.; Deng, C.-L.; Liu, S.-Q.; Zhang, H.-L.; Shang, B.-D.; Shi, P.-Y.; Yuan, Z.-M.; Zhang, B. Genetic interaction between NS4A and NS4B for replication of Japanese encephalitis virus. J. Gen. Virol. 2015, 96, 1264–1275. [Google Scholar] [CrossRef]
- Miller, S.; Kastner, S.; Krijnse-Locker, J.; Bühler, S.; Bartenschlager, R. The Non-structural Protein 4A of Dengue Virus Is an Integral Membrane Protein Inducing Membrane Alterations in a 2K-regulated Manner. J. Biol. Chem. 2007, 282, 8873–8882. [Google Scholar] [CrossRef] [Green Version]
- Teo, C.S.H.; Chu, J.J.H. Cellular Vimentin Regulates Construction of Dengue Virus Replication Complexes through Interaction with NS4A Protein. J. Virol. 2014, 88, 1897–1913. [Google Scholar] [CrossRef] [Green Version]
- Kaufusi, P.H.; Kelley, J.F.; Yanagihara, R.; Nerurkar, V.R. Induction of Endoplasmic Reticulum-Derived Replication-Competent Membrane Structures by West Nile Virus Non-Structural Protein 4B. PLoS ONE 2014, 9, e84040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, S.; Sparacio, S.; Bartenschlager, R. Subcellular Localization and Membrane Topology of the Dengue Virus Type 2 Non-structural Protein 4B. J. Biol. Chem. 2006, 281, 8854–8863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roosendaal, J.; Westaway, E.G.; Khromykh, A.; MacKenzie, J.M. Regulated Cleavages at the West Nile Virus NS4A-2K-NS4B Junctions Play a Major Role in Rearranging Cytoplasmic Membranes and Golgi Trafficking of the NS4A Protein. J. Virol. 2006, 80, 4623–4632. [Google Scholar] [CrossRef] [Green Version]
- Shiryaev, S.A.; Chernov, A.V.; Aleshin, A.E.; Shiryaeva, T.N.; Strongin, A.Y. NS4A regulates the ATPase activity of the NS3 helicase: A novel cofactor role of the non-structural protein NS4A from West Nile virus. J. Gen. Virol. 2009, 90, 2081–2085. [Google Scholar] [CrossRef]
- Youn, S.; Li, T.; McCune, B.T.; Edeling, M.A.; Fremont, D.H.; Cristea, I.M.; Diamond, M.S. Evidence for a Genetic and Physical Interaction between Nonstructural Proteins NS1 and NS4B That Modulates Replication of West Nile Virus. J. Virol. 2012, 86, 7360–7371. [Google Scholar] [CrossRef] [Green Version]
- Lindenbach, B.D.; Rice, C.M. Genetic Interaction of Flavivirus Nonstructural Proteins NS1 and NS4A as a Determinant of Replicase Function. J. Virol. 1999, 73, 4611–4621. [Google Scholar] [CrossRef] [Green Version]
- Aktepe, T.E.; Liebscher, S.; Prier, J.; Simmons, C.P.; Mackenzie, J.M. The Host Protein Reticulon 3.1A Is Utilized by Flaviviruses to Facilitate Membrane Remodelling. Cell Rep. 2017, 21, 1639–1654. [Google Scholar] [CrossRef] [Green Version]
- Rismanchi, N.; Soderblom, C.; Stadler, J.; Zhu, P.-P.; Blackstone, C. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum. Mol. Genet. 2008, 17, 1591–1604. [Google Scholar] [CrossRef]
- Hu, J.; Rapoport, T.A. Fusion of the endoplasmic reticulum by membrane-bound GTPases. Semin. Cell Dev. Biol. 2016, 60, 105–111. [Google Scholar] [CrossRef]
- Monel, B.; Rajah, M.M.; Hafirassou, M.L.; Ahmed, S.S.; Burlaud-Gaillard, J.; Zhu, P.-P.; Nevers, Q.; Buchrieser, J.; Porrot, F.; Meunier, C.; et al. Atlastin Endoplasmic Reticulum-Shaping Proteins Facilitate Zika Virus Replication. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Tabata, K.; Arimoto, M.; Arakawa, M.; Nara, A.; Saito, K.; Omori, H.; Arai, A.; Ishikawa, T.; Konishi, E.; Suzuki, R.; et al. Unique Requirement for ESCRT Factors in Flavivirus Particle Formation on the Endoplasmic Reticulum. Cell Rep. 2016, 16, 2339–2347. [Google Scholar] [CrossRef] [Green Version]
- Hurley, J.H. ESCRTs are everywhere. EMBO J. 2015, 34, 2398–2407. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, M.; Chen, J.; Janda, M.; Sullivan, M.; Boon, J.D.; Ahlquist, P. A Positive-Strand RNA Virus Replication Complex Parallels Form and Function of Retrovirus Capsids. Mol. Cell 2002, 9, 505–514. [Google Scholar] [CrossRef]
- Diaz, A.; Wang, X.; Ahlquist, P. Membrane-shaping host reticulon proteins play crucial roles in viral RNA replication compartment formation and function. Proc. Natl. Acad. Sci. USA 2010, 107, 16291–16296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diaz, A.; Zhang, J.; Ollwerther, A.; Wang, X.; Ahlquist, P. Host ESCRT proteins are required for bromovirus RNA replication compartment assembly and function. PLoS Pathog. 2015, 11, e1004742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barajas, D.; Jiang, Y.; Nagy, P.D. A Unique Role for the Host ESCRT Proteins in Replication of Tomato bushy stunt virus. PLoS Pathog. 2009, 5, e1000705. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar] [CrossRef]
- Sharma, M.; Bhattacharyya, S.; Sharma, K.B.; Chauhan, S.; Asthana, S.; Abdin, M.Z.; Vrati, S.; Kalia, M. Japanese encephalitis virus activates autophagy through XBP1 and ATF6 ER stress sensors in neuronal cells. J. Gen. Virol. 2017, 98, 1027–1039. [Google Scholar] [CrossRef]
- Shives, K.D.; Beatman, E.L.; Chamanian, M.; O’Brien, C.; Hobson-Peters, J.; Beckham, J.D. West Nile Virus-Induced Activation of Mammalian Target of Rapamycin Complex 1 Supports Viral Growth and Viral Protein Expression. J. Virol. 2014, 88, 9458–9471. [Google Scholar] [CrossRef] [Green Version]
- Heaton, N.S.; Randall, G. Dengue Virus-Induced Autophagy Regulates Lipid Metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef] [Green Version]
- Liang, Q.; Luo, Z.; Zeng, J.; Chen, W.; Foo, S.S.; Lee, S.A.; Ge, J.; Wang, S.; Goldman, S.A.; Zlokovic, B.V.; et al. Zika Virus NS4A and NS4B proteins deregulate Akt-mTOR signaling in human fetal neural stem cells to inhibit neurogenesis and induce autophagy. Cell Stem Cell 2016, 19, 663–671. [Google Scholar] [CrossRef] [Green Version]
- Chiramel, A.I.; Best, S.M. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. 2018, 254, 34–40. [Google Scholar] [CrossRef]
- Metz, P.; Chiramel, A.; Chatel-Chaix, L.; Alvisi, G.; Bankhead, P.; Mora-Rodríguez, R.; Long, G.; Hamacher-Brady, A.; Brady, N.R.; Bartenschlager, R. Dengue Virus Inhibition of Autophagic Flux and Dependency of Viral Replication on Proteasomal Degradation of the Autophagy Receptor p62. J. Virol. 2015, 89, 8026–8041. [Google Scholar] [CrossRef] [Green Version]
- Beatman, E.; Oyer, R.; Shives, K.D.; Hedman, K.; Brault, A.C.; Tyler, K.L.; Beckham, J.D. West Nile virus growth is independent of autophagy activation. Virology 2012, 433, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McLean, J.E.; Wudzinska, A.; Datan, E.; Quaglino, D.; Zakeri, Z. Flavivirus NS4A-induced Autophagy Protects Cells against Death and Enhances Virus Replication. J. Biol. Chem. 2011, 286, 22147–22159. [Google Scholar] [CrossRef] [Green Version]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, A.S.; Lennemann, N.J.; Coyne, C.B. BPIFB3 Regulates Endoplasmic Reticulum Morphology to Facilitate Flavivirus Replication. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Liu, Y.; Gordesky-Gold, B.; Leney-Greene, M.; Weinbren, N.L.; Tudor, M.; Cherry, S. Inflammation-Induced, STING-Dependent Autophagy Restricts Zika Virus Infection in the Drosophila Brain. Cell Host Microbe 2018, 24, 57–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennemann, N.J.; Coyne, C.B. Dengue and Zika viruses subvert reticulophagy by NS2B3-mediated cleavage of FAM134B. Autophagy 2017, 13, 322–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, H.-H.; Schneider, W.M.; Rozen-Gagnon, K.; Miles, L.A.; Schuster, F.; Razooky, B.; Jacobson, E.; Wu, X.; Yi, S.; Rudin, C.M.; et al. TMEM41B Is a Pan-Flavivirus Host Factor. Cell 2021, 184, 133–148.e20. [Google Scholar] [CrossRef]
- Morita, K.; Hama, Y.; Izume, T.; Tamura, N.; Ueno, T.; Yamashita, Y.; Sakamaki, Y.; Mimura, K.; Morishita, H.; Shihoya, W.; et al. Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. J. Cell Biol. 2018, 217, 3817–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.E.; Wang, Y.; Du, X.; Zhang, T.; Mak, H.Y.; Hancock, S.E.; McEwen, H.; Pandzic, E.; Whan, R.M.; Aw, Y.C.; et al. TMEM41B and VMP1 are scramblases and regulate the distribution of cholesterol and phosphatidylserine. J. Cell Biol. 2021, 220. [Google Scholar] [CrossRef] [PubMed]
- Martín-Acebes, M.A.; Merino-Ramos, T.; Blázquez, A.-B.; Casas, J.; Escribano-Romero, E.; Sobrino, F.; Saiz, J.-C. The Composition of West Nile Virus Lipid Envelope Unveils a Role of Sphingolipid Metabolism in Flavivirus Biogenesis. J. Virol. 2014, 88, 12041–12054. [Google Scholar] [CrossRef] [Green Version]
- Reiss, S.; Rebhan, I.; Backes, P.; Romero-Brey, I.; Erfle, H.; Matula, P.; Kaderali, L.; Poenisch, M.; Blankenburg, H.; Hiet, M.-S.; et al. Recruitment and Activation of a Lipid Kinase by Hepatitis C Virus NS5A Is Essential for Integrity of the Membranous Replication Compartment. Cell Host Microbe 2011, 9, 32–45. [Google Scholar] [CrossRef] [Green Version]
- Hsu, N.-Y.; Ilnytska, O.; Belov, G.; Santiana, M.; Chen, Y.-H.; Takvorian, P.M.; Pau, C.; Van Der Schaar, H.; Kaushik-Basu, N.; Balla, T.; et al. Viral Reorganization of the Secretory Pathway Generates Distinct Organelles for RNA Replication. Cell 2010, 141, 799–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amako, Y.; Sarkeshik, A.; Hotta, H.; Yates, J., III; Siddiqui, A. Role of oxysterol binding protein in hepatitis C virus infection. J. Virol. 2009, 83, 9237–9246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Perry, J.W.; Lauring, A.S.; Neddermann, P.; De Francesco, R.; Tai, A.W. Oxysterol-Binding Protein Is a Phosphatidylinositol 4-Kinase Effector Required for HCV Replication Membrane Integrity and Cholesterol Trafficking. Gastroenterology 2014, 146, 1373–1385.e11. [Google Scholar] [CrossRef]
- Soto-Acosta, R.; Bautista-Carbajal, P.; Cervantes-Salazar, M.; Angel-Ambrocio, A.H.; Del Angel, R.M. DENV up-regulates the HMG-CoA reductase activity through the impairment of AMPK phosphorylation: A potential antiviral target. PLoS Pathog. 2017, 13, e1006257. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Perera, R.; Berger, K.L.; Khadka, S.; LaCount, D.J.; Kuhn, R.J.; Randall, G. Dengue virus nonstructural protein 3 redistributes fatty acid synthase to sites of viral replication and increases cellular fatty acid synthesis. Proc. Natl. Acad. Sci. USA 2010, 107, 17345–17350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthoux, L. The Restrictome of Flaviviruses. Virol. Sin. 2020, 35, 363–377. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Chan, Y.K.; Gack, M.U. Viral evasion of intracellular DNA and RNA sensing. Nat. Rev. Genet. 2016, 14, 360–373. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, O.; Akira, S. Pattern Recognition Receptors and Inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nazmi, A.; Dutta, K.; Hazra, B.; Basu, A. Role of pattern recognition receptors in flavivirus infections. Virus Res. 2014, 185, 32–40. [Google Scholar] [CrossRef]
- Loo, Y.-M.; Fornek, J.; Crochet, N.; Bajwa, G.; Perwitasari, O.; Martinez-Sobrido, L.; Akira, S.; Gill, M.A.; García-Sastre, A.; Katze, M.G.; et al. Distinct RIG-I and MDA5 Signaling by RNA Viruses in Innate Immunity. J. Virol. 2007, 82, 335–345. [Google Scholar] [CrossRef] [Green Version]
- Nasirudeen, A.M.A.; Wong, H.H.; Thien, P.; Xu, S.; Lam, K.-P.; Liu, D.X. RIG-I, MDA5 and TLR3 Synergistically Play an Important Role in Restriction of Dengue Virus Infection. PLoS Negl. Trop. Dis. 2011, 5, e926. [Google Scholar] [CrossRef]
- Fredericksen, B.L.; Keller, B.C.; Fornek, J.; Katze, M.G.; Gale, M. Establishment and Maintenance of the Innate Antiviral Response to West Nile Virus Involves both RIG-I and MDA5 Signaling through IPS-1. J. Virol. 2007, 82, 609–616. [Google Scholar] [CrossRef] [Green Version]
- Welte, T.; Reagan, K.; Fang, H.; Machain-Williams, C.; Zheng, X.; Mendell, N.; Chang, G.-J.J.; Wu, P.; Blair, C.D.; Wang, T. Toll-like receptor 7-induced immune response to cutaneous West Nile virus infection. J. Gen. Virol. 2009, 90, 2660–2668. [Google Scholar] [CrossRef]
- Maringer, K.; Fernandez-Sesma, A. Message in a bottle: Lessons learned from antagonism of STING signalling during RNA virus infection. Cytokine Growth Factor Rev. 2014, 25, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Moretti, J.; Roy, S.; Bozec, D.; Martinez, J.; Chapman, J.R.; Ueberheide, B.; Lamming, D.W.; Chen, Z.; Horng, T.; Yeretssian, G.; et al. STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell 2017, 171, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-Y.; Chang, T.-H.; Liang, J.-J.; Chiang, R.-L.; Lee, Y.-L.; Liao, C.-L.; Lin, Y.-L. Dengue Virus Targets the Adaptor Protein MITA to Subvert Host Innate Immunity. PLoS Pathog. 2012, 8, e1002780. [Google Scholar] [CrossRef]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV Inhibits Type I IFN Production in Infected Cells by Cleaving Human STING. PLoS Pathog. 2012, 8, e1002934. [Google Scholar] [CrossRef] [Green Version]
- Diamond, M.S. Mechanisms of Evasion of the Type I Interferon Antiviral Response by Flaviviruses. J. Interf. Cytokine Res. 2009, 29, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-Stimulated Genes: A Complex Web of Host Defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef]
- Hornung, V.; Hartmann, R.; Ablasser, A.; Hopfner, K.-P. OAS proteins and cGAS: Unifying concepts in sensing and responding to cytosolic nucleic acids. Nat. Rev. Immunol. 2014, 14, 521–528. [Google Scholar] [CrossRef] [PubMed]
- Silverman, R.H. Viral Encounters with 2′,5′-Oligoadenylate Synthetase and RNase L during the Interferon Antiviral Response. J. Virol. 2007, 81, 12720–12729. [Google Scholar] [CrossRef] [Green Version]
- Perelygin, A.A.; Scherbik, S.V.; Zhulin, I.; Stockman, B.M.; Li, Y.; Brinton, M.A. Positional cloning of the murine flavivirus resistance gene. Proc. Natl. Acad. Sci. USA 2002, 99, 9322–9327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R.-J.; Yu, H.-P.; Chang, B.-L.; Tang, W.-C.; Liao, C.-L.; Lin, Y.-L. Distinct Antiviral Roles for Human 2′,5′-Oligoadenylate Synthetase Family Members against Dengue Virus Infection. J. Immunol. 2009, 183, 8035–8043. [Google Scholar] [CrossRef] [Green Version]
- Whelan, J.N.; Li, Y.; Silverman, R.H.; Weiss, S.R. Zika Virus Production Is Resistant to RNase L Antiviral Activity. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Helbig, K.J.; Beard, M.R. The Role of Viperin in the Innate Antiviral Response. J. Mol. Biol. 2014, 426, 1210–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghosh, S.; Marsh, E.N.G. Viperin: An ancient radical SAM enzyme finds its place in modern cellular metabolism and innate immunity. J. Biol. Chem. 2020, 295, 11513–11528. [Google Scholar] [CrossRef]
- Seo, J.-Y.; Yaneva, R.; Cresswell, P. Viperin: A Multifunctional, Interferon-Inducible Protein that Regulates Virus Replication. Cell Host Microbe 2011, 10, 534–539.e3. [Google Scholar] [CrossRef] [Green Version]
- Fenwick, M.K.; Li, Y.; Cresswell, P.; Modis, Y.; Ealick, S.E. Structural studies of viperin, an antiviral radical SAM enzyme. Proc. Natl. Acad. Sci. USA 2017, 114, 6806–6811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gizzi, A.S.; Grove, T.L.; Arnold, J.J.; Jose, J.; Jangra, R.K.; Garforth, S.J.; Du, Q.; Cahill, S.M.; Dulyaninova, N.G.; Love, J.D.; et al. A naturally occurring antiviral ribonucleotide encoded by the human genome. Nature 2018, 558, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Helbig, K.J.; Carr, J.M.; Calvert, J.K.; Wati, S.; Clarke, J.N.; Eyre, N.S.; Narayana, S.K.; Fiches, G.N.; McCartney, E.M.; Beard, M.R. Viperin Is Induced following Dengue Virus Type-2 (DENV-2) Infection and Has Anti-viral Actions Requiring the C-terminal End of Viperin. PLoS Neglected Trop. Dis. 2013, 7, e2178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, N.; Porter, A.C.G. Identification of a novel gene family that includes the interferon-inducible human genes 6–16 and ISG12. BMC Genom. 2004, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gjermandsen, I.M.; Justesen, J.; Martensen, P.M. The Interferon-Induced Gene ISG12 Is Regulated by Various Cytokines as the Gene 6-16 In Human Cell Lines. Cytokine 2000, 12, 233–238. [Google Scholar] [CrossRef]
- Qi, Y.; Li, Y.; Zhang, Y.; Zhang, L.; Wang, Z.; Zhang, X.; Gui, L.; Huang, J. IFI6 inhibits apoptosis via mitochondrial-dependent pathway in dengue virus 2 infected vascular endothelial cells. PLoS ONE 2015, 10, e0132743. [Google Scholar]
- Park, G.-H.; Kim, K.-Y.; Cho, S.W.; Cheong, J.Y.; Yu, G.I.; Shin, D.H.; Kwack, K.B. Association between Interferon-Inducible Protein 6 (IFI6) Polymorphisms and Hepatitis B Virus Clearance. Genom. Inform. 2013, 11, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szegedi, K.; Sonkoly, E.; Nagy, N.; Németh, I.B.; Bata-Csörgő, Z.; Kemény, L.; Dobozy, A.; Széll, M. The anti-apoptotic protein G1P3 is overexpressed in psoriasis and regulated by the non-coding RNA, PRINS. Exp. Dermatol. 2010, 19, 269–278. [Google Scholar] [CrossRef]
- Li, J.; Huang, K.; Hu, G.; Babarinde, I.A.; Li, Y.; Dong, X.; Chen, Y.-S.; Shang, L.; Guo, W.; Wang, J.; et al. An alternative CTCF isoform antagonizes canonical CTCF occupancy and changes chromatin architecture to promote apoptosis. Nat. Commun. 2019, 10, 1535. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Ding, S.C.; Cho, H.; Chung, B.C.; Gale, M.; Chanda, S.K.; Diamond, M.S. A Short Hairpin RNA Screen of Interferon-Stimulated Genes Identifies a Novel Negative Regulator of the Cellular Antiviral Response. mBio 2013, 4, e00385-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dukhovny, A.; Lamkiewicz, K.; Chen, Q.; Fricke, M.; Jabrane-Ferrat, N.; Marz, M.; Jung, J.U.; Sklan, E.H. A CRISPR Activation Screen Identifies Genes That Protect against Zika Virus Infection. J. Virol. 2019, 93, e00211-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W.; Dorner, M.; Feulner, M.; Imanaka, N.; Murphy, M.Y.; Ploss, A.; Rice, C.M. Dengue reporter viruses reveal viral dynamics in interferon receptor-deficient mice and sensitivity to interferon effectors in vitro. Proc. Natl. Acad. Sci. USA 2012, 109, 14610–14615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, R.B.; Ohlson, M.B.; Eitson, J.L.; Kumar, A.; McDougal, M.B.; Boys, I.N.; Mar, K.B.; De La Cruz-Rivera, P.C.; Douglas, C.; Konopka, G.; et al. A CRISPR screen identifies IFI6 as an ER-resident interferon effector that blocks flavivirus replication. Nat. Microbiol. 2018, 3, 1214–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Gent, M.; Sparrer, K.M.; Gack, M.U. TRIM Proteins and Their Roles in Antiviral Host Defenses. Annu. Rev. Virol. 2018, 5, 385–405. [Google Scholar] [CrossRef]
- Taylor, R.T.; Lubick, K.J.; Robertson, S.J.; Broughton, J.P.; Bloom, M.E.; Bresnahan, W.A.; Best, S.M. TRIM79α, an Interferon-Stimulated Gene Product, Restricts Tick-Borne Encephalitis Virus Replication by Degrading the Viral RNA Polymerase. Cell Host Microbe 2011, 10, 185–196. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, Y.; Chin, W.-X.; Han, Q.; Ichiyama, K.; Lee, C.H.; Eyo, Z.W.; Ebina, H.; Takahashi, H.; Takahashi, C.; Tan, B.H.; et al. Characterization of RyDEN (C19orf66) as an Interferon-Stimulated Cellular Inhibitor against Dengue Virus Replication. PLoS Pathog. 2016, 12, e1005357. [Google Scholar] [CrossRef] [Green Version]
- Balinsky, C.A.; Schmeisser, H.; Wells, A.; Ganesan, S.; Jin, T.; Singh, K.; Zoon, K.C. IRAV (FLJ11286), an Interferon-Stimulated Gene with Antiviral Activity against Dengue Virus, Interacts with MOV10. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Yang, X.; Yao, Z.; Dong, X.; Zhang, D.; Hu, Y.; Zhang, S.; Lin, J.; Chen, J.; An, S.; et al. C19orf66 interrupts Zika virus replication by inducing lysosomal degradation of viral NS3. PLoS Negl. Trop. Dis. 2020, 14, e0008083. [Google Scholar] [CrossRef] [Green Version]
- Boys, I.N.; Xu, E.; Mar, K.B.; De La Cruz-Rivera, P.C.; Eitson, J.L.; Moon, B.; Schoggins, J.W. RTP4 Is a Potent IFN-Inducible Anti-flavivirus Effector Engaged in a Host-Virus Arms Race in Bats and Other Mammals. Cell Host Microbe 2020, 28, 712–723.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xuan, Y.; Han, Y.; Ding, X.; Ye, K.; Yang, F.; Gao, P.; Goff, S.P.; Gao, G. Regulation of HIV-1 Gag-Pol Expression by Shiftless, an Inhibitor of Programmed -1 Ribosomal Frameshifting. Cell 2019, 176, 625–635.e14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, W.; Srivastav, K.; Muller, M. C19ORF66 Broadly Escapes Virus-Induced Endonuclease Cleavage and Restricts Kaposi’s Sarcoma-Associated Herpesvirus. J. Virol. 2019, 93, e00373-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, A.J.; Thoreen, C.C.; Dedeic, Z.; Chettle, J.; Roux, P.P.; Blagden, S.P. Controversies around the function of LARP1. RNA Biol. 2021, 18, 207–217. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morita, E.; Suzuki, Y. Membrane-Associated Flavivirus Replication Complex—Its Organization and Regulation. Viruses 2021, 13, 1060. https://doi.org/10.3390/v13061060
Morita E, Suzuki Y. Membrane-Associated Flavivirus Replication Complex—Its Organization and Regulation. Viruses. 2021; 13(6):1060. https://doi.org/10.3390/v13061060
Chicago/Turabian StyleMorita, Eiji, and Youichi Suzuki. 2021. "Membrane-Associated Flavivirus Replication Complex—Its Organization and Regulation" Viruses 13, no. 6: 1060. https://doi.org/10.3390/v13061060
APA StyleMorita, E., & Suzuki, Y. (2021). Membrane-Associated Flavivirus Replication Complex—Its Organization and Regulation. Viruses, 13(6), 1060. https://doi.org/10.3390/v13061060