
Full-Genome Sequences of Alphacoronaviruses and Astroviruses from Myotis and Pipistrelle Bats in Denmark


Abstract
:1. Introduction
2. Materials and Methods
3. Results
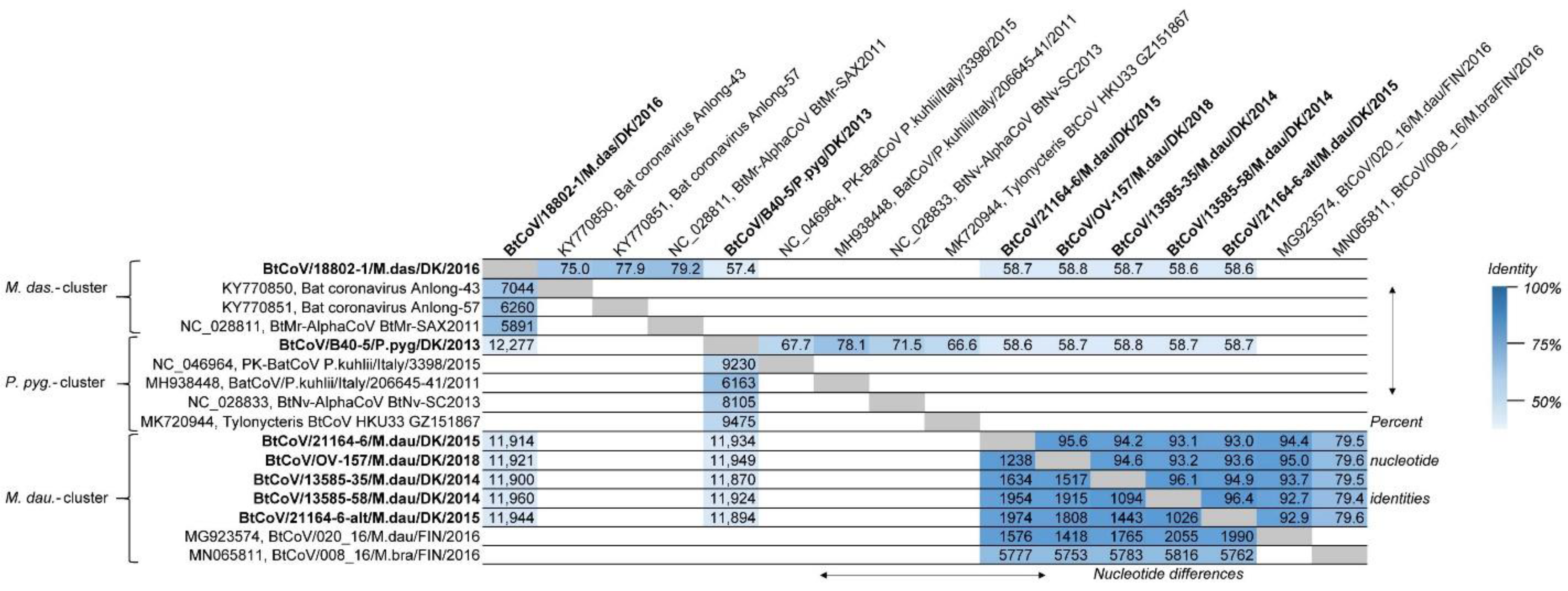
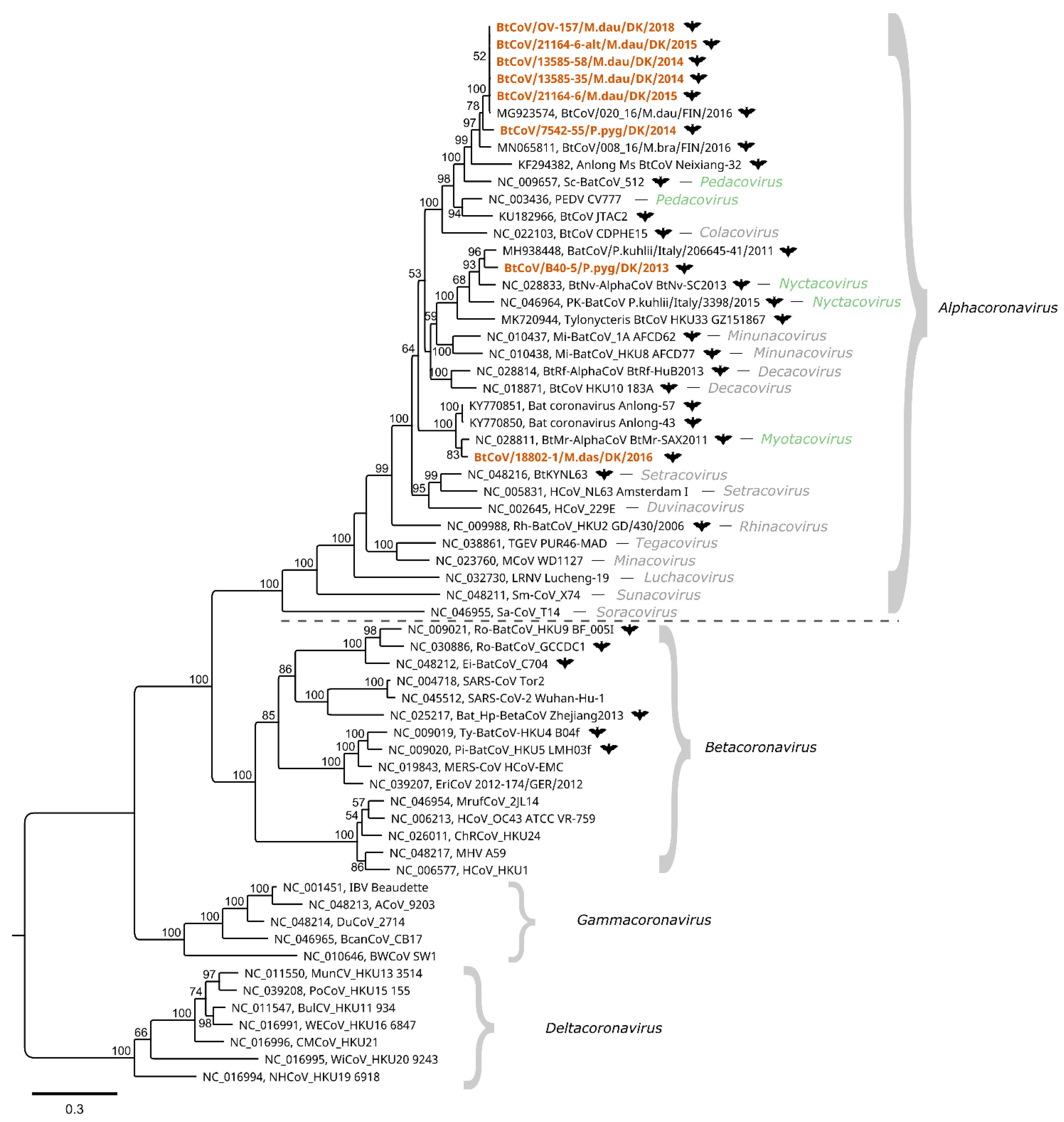
3.1. Bat Coronaviruses
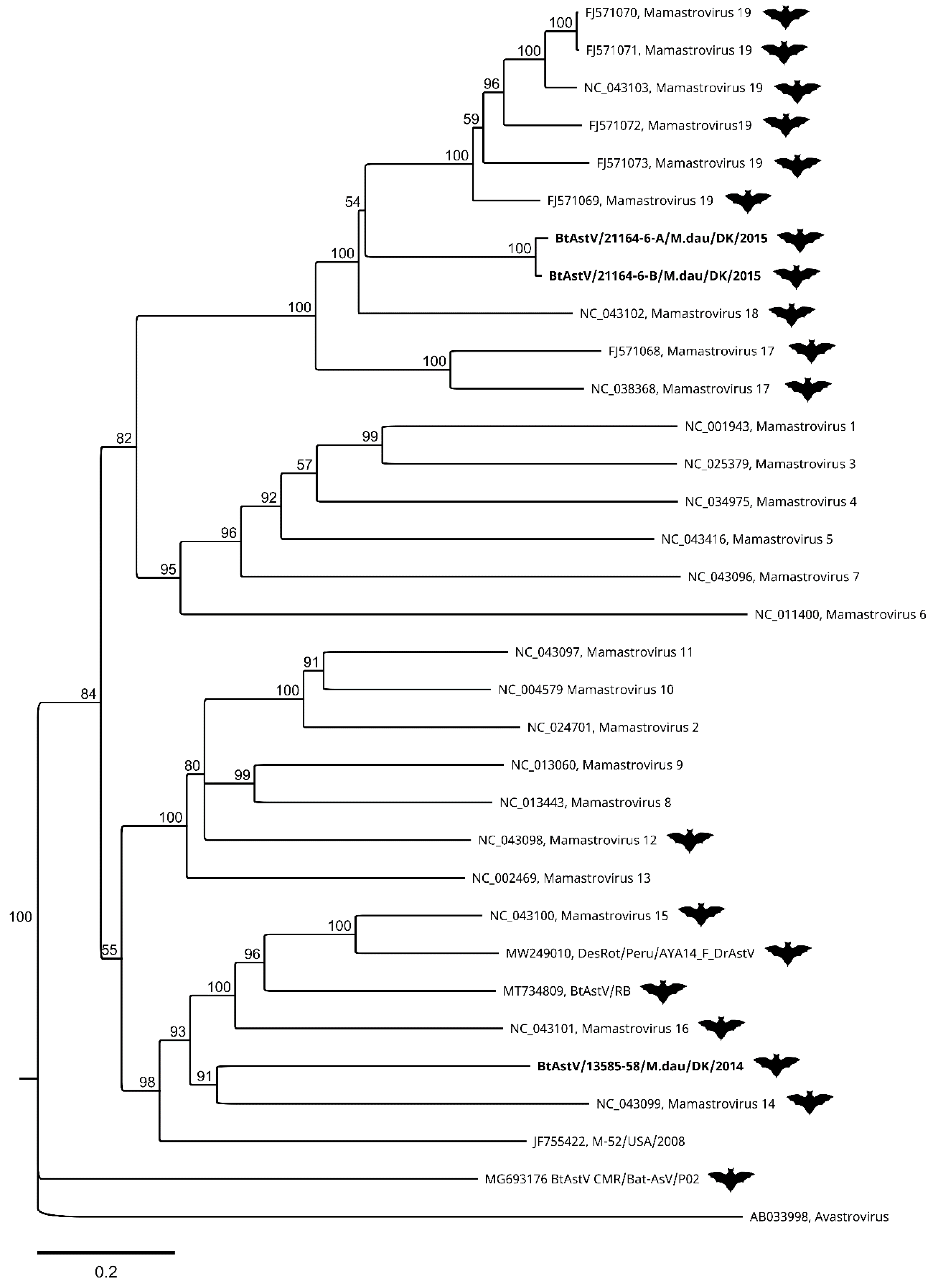
3.2. Bat Astroviruses
3.3. Rhopalosiphum Padi Viruses
3.4. Kadipiro Viruses
3.5. Other Viral and Microbial Agents Present in the Data
4. Discussion
Supplementary Materials
Author Contributions
Funding
Ethical approval
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burgin, C.J.; Colella, J.P.; Kahn, P.L.; Upham, N.S. How many species of mammals are there? J. Mammal. 2018, 99, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kunz, T.H.; de Torrez, E.B.; Bauer, D.; Lobova, T.; Fleming, T.H. Ecosystem services provided by bats. Ann. N. Y. Acad. Sci. 2011, 1223, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Baagøe, H.J. Flagermus, Chiroptera. In Dansk Pattedyratlas, 1st ed.; Baagøe, H.J., Jensen, T.S., Eds.; Gyldendal: Copenhagen, Denmark, 2007; pp. 38–98. [Google Scholar]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-Borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Subudhi, S.; Rapin, N.; Misra, V. Immune system modulation and viral persistence in bats: Understanding viral spillover. Viruses 2019, 11, 192. [Google Scholar] [CrossRef] [Green Version]
- Schountz, T.; Baker, M.L.; Butler, J.; Munster, V. Immunological control of viral infections in bats and the emergence of viruses highly pathogenic to humans. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.L. Emerging infectious diseases associated with bat viruses. Sci. China Life Sci. 2013, 56, 678–682. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.; Lau, S.; Woo, P.; Yuen, K.Y. Bats as a continuing source of emerging infections in humans. Rev. Med. Virol. 2007, 17, 67–91. [Google Scholar] [CrossRef]
- Chen, L.; Liu, B.; Yang, J.; Jin, Q. DBatVir: The database of bat-associated viruses. Database 2014, 2014, bau021. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Victoria, J.G.; Wang, C.; Jones, M.; Fellers, G.M.; Kunz, T.H.; Delwart, E. Bat guano virome: Predominance of dietary viruses from insects and plants plus novel mammalian viruses. J. Virol. 2010, 84, 6955–6965. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J.; et al. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef] [Green Version]
- Hu, D.; Zhu, C.; Wang, Y.; Ai, L.; Yang, L.; Ye, F.; Ding, C.; Chen, J.; He, B.; Zhu, J.; et al. Virome analysis for identification of novel mammalian viruses in bats from Southeast China. Sci. Rep. 2017, 7, 10917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donaldson, E.F.; Haskew, A.N.; Gates, J.E.; Huynh, J.; Moore, C.J.; Frieman, M.B. Metagenomic analysis of the viromes of three North American bat species: Viral diversity among different bat species that share a common habitat. J. Virol. 2010, 84, 13004–13018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Altan, E.; Reyes, G.; Halstead, B.; Deng, X.; Delwart, E. Virome of bat guano from nine Northern California roosts. J. Virol. 2021, 95. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Groot, R.J.; Baker, S.C.; Baric, R.; Enjuanes, L.; Gorbalenya, A.E.; Holmes, K.V.; Perlman, S.; Poon, L.; Rottier, P.J.M.; Talbot, P.J.; et al. Family: Coronaviridae. In Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 806–828. [Google Scholar] [CrossRef]
- Woo, P.C.Y.; Lau, S.K.P.; Lam, C.S.F.; Lau, C.C.Y.; Tsang, A.K.L.; Lau, J.H.N.; Bai, R.; Teng, J.L.L.; Tsang, C.C.C.; Wang, M.; et al. Discovery of seven novel mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavi. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorbalenya, A.E.; Baker, S.C.; Baric, R.S.; de Groot, R.J.; Drosten, C.; Gulyaeva, A.A.; Haagmans, B.L.; Lauber, C.; Leontovich, A.M.; Neuman, B.W.; et al. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. 2020, 30, 2196–2203. [Google Scholar] [CrossRef]
- Murakami, S.; Kitamura, T.; Suzuki, J.; Sato, R.; Aoi, T.; Fujii, M.; Matsugo, H.; Kamiki, H.; Ishida, H.; Takenaka-Uema, A.; et al. Detection and characterization of bat sarbecovirus phylogenetically related to SARS-CoV-2, Japan. Emerg. Infect. Dis. 2020, 26, 3025–3029. [Google Scholar] [CrossRef]
- Wacharapluesadee, S.; Tan, C.W.; Maneeorn, P.; Duengkae, P.; Zhu, F.; Joyjinda, Y.; Kaewpom, T.; Chia, W.N.; Ampoot, W.; Lim, B.L.; et al. Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 2021, 12. [Google Scholar] [CrossRef]
- Hul, V.; Delaune, D.; Karlsson, E.A.; Hassanin, A.; Tey, P.O.; Baidaliuk, A.; Gámbaro, F.; Tu, V.T.; Keatts, L.; Mazet, J.; et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. bioRxiv 2021. [Google Scholar] [CrossRef]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and sources of endemic human coronaviruses. Adv. Virus Res. 2018, 100, 163–188. [Google Scholar] [CrossRef] [PubMed]
- Pfefferle, S.; Oppong, S.; Drexler, J.F.; Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Müller, M.A.; Annan, A.; Vallo, P.; Adu-Sarkodie, Y.; et al. Distant relatives of severe acute respiratory syndrome coronavirus and close relatives of human coronavirus 229E in bats, Ghana. Emerg. Infect. Dis. 2009, 15, 1377–1384. [Google Scholar] [CrossRef]
- Wang, W.; Lin, X.D.; Guo, W.P.; Zhou, R.H.; Wang, M.R.; Wang, C.Q.; Ge, S.; Mei, S.H.; Li, M.H.; Shi, M.; et al. Discovery, diversity and evolution of novel coronaviruses sampled from rodents in China. Virology 2015, 474, 19–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lazov, C.M.; Chriél, M.; Baagøe, H.J.; Fjederholt, E.; Deng, Y.; Kooi, E.A.; Belsham, G.J.; Bøtner, A.; Rasmussen, T.B. Detection and characterization of distinct alphacoronaviruses in five different bat species in Denmark. Viruses 2018, 10, 486. [Google Scholar] [CrossRef] [Green Version]
- Kivistö, I.; Tidenberg, E.-M.; Lilley, T.; Suominen, K.; Forbes, K.M.; Vapalahti, O.; Huovilainen, A.; Sironen, T. First report of coronaviruses in Northern European bats. Vector Borne Zoonotic Dis. 2019, 20, 155–158. [Google Scholar] [CrossRef] [PubMed]
- Moon, J.S.; Domier, L.L.; McCoppin, N.K.; D’Arcy, C.J.; Jin, H. Nucleotide sequence analysis shows that Rhopalosiphum padi virus is a member of a novel group of insect-infecting RNA viruses. Virology 1998, 243, 54–65. [Google Scholar] [CrossRef] [Green Version]
- Attoui, H.; Billoir, F.; Biagini, P.; De Micco, P.; De Lamballerie, X. Complete sequence determination and genetic analysis of Banna virus and Kadipiro virus: Proposal for assignment to a new genus (Seadornavirus) within the family Reoviridae. J. Gen. Virol. 2000, 81, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Bosch, A.; Guix, S.; Krishna, N.K.; Méndez, E.; Monroe, S.S.; Pantin-Jackwood, M.; Schultz-Cherry, S. Family: Astroviridae. In Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 953–959. [Google Scholar] [CrossRef]
- Fischer, K.; dos Reis, V.P.; Balkema-Buschmann, A. Bat astroviruses: Towards understanding the transmission dynamics of a neglected virus family. Viruses 2017, 9, 34. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, S. FastQC v. 0.11.8. Babraham Institute Bioinformatics Group. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 25 April 2021).
- Geneious Prime v. 2021.1.1 Biomatters Ltd. Available online: https://www.geneious.com (accessed on 25 April 2021).
- Bushnell, B. BBDuk Trimmer v. 1.0. Biomatters Ltd. Available online: https://www.geneious.com/plugins/bbduk/#quick-start (accessed on 25 April 2021).
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [Green Version]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef]
- ICTV Master Species List 2019.v1. Available online: https://talk.ictvonline.org/files/master-species-lists/m/msl/9601 (accessed on 25 April 2021).
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.C.E.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Reorganization and Expansion of the Order Nidovirales at the Family and Sub-Order Ranks, Code 2017.015S. Available online: https://talk.ictvonline.org/ictv/proposals/2017.012_015S.A.v1.Nidovirales.zip (accessed on 25 April 2021).
- Fischer, K.; Zeus, V.; Kwasnitschka, L.; Kerth, G.; Haase, M.; Groschup, M.H.; Balkema-Buschmann, A. Insectivorous bats carry host specific astroviruses and coronaviruses across different regions in Germany. Infect. Genet. Evol. 2016, 37, 108–116. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Poon, L.L.M.; Guan, Y.; Peiris, J.S.M. Novel astroviruses in insectivorous bats. J. Virol. 2008, 82, 9107–9114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- In the Genus Mamastrovirus, Family Astroviridae, Create 14 Species, Abolish One Species and Rename 5 Species, Code 2010.018a-cV. Available online: https://talk.ictvonline.org/ictv/proposals/2010.018a-cV.A.v4.Mamastrovirus.pdf (accessed on 25 April 2021).
- Jones, R.A.C. Plant and insect viruses in managed and natural environments: Novel and neglected transmission pathways. In Advances in Virus Research, 1st ed.; Elsevier Inc.: Amsterdam, The Netherlands, 2018. [Google Scholar] [CrossRef]
- Terenin, I.M.; Dmitriev, S.E.; Andreev, D.E.; Royall, E.; Belsham, G.J.; Roberts, L.O.; Shatsky, I.N. A cross-kingdom internal ribosome entry site reveals a simplified mode of internal ribosome entry. Mol. Cell. Biol. 2005, 25, 7879–7888. [Google Scholar] [CrossRef] [Green Version]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guérin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E. ICTV virus taxonomy profile: Dicistroviridae. J. Gen. Virol. 2017, 98, 355–356. [Google Scholar] [CrossRef]
- Zana, B.; Kemenesi, G.; Urbán, P.; Földes, F.; Görföl, T.; Estók, P.; Boldogh, S.; Kurucz, K.; Jakab, F. Metagenomic analysis of bat guano samples revealed the presence of viruses potentially carried by insects, among others by Apis mellifera in Hungary. Acta Vet. Hung. 2018, 66, 151–161. [Google Scholar] [CrossRef] [PubMed]
- Ban, L.; Didon, A.; Jonsson, L.M.V.; Glinwood, R.; Delp, G. An improved detection method for the Rhopalosiphum padi virus (RhPV) allows monitoring of its presence in aphids and movement within plants. J. Virol. Methods 2007, 142, 136–142. [Google Scholar] [CrossRef]
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Dempsey, D.M.; Dutilh, B.E.; Harrach, B.; Harrison, R.L.; Hendrickson, R.C.; et al. Changes to virus taxonomy and the statutes ratified by the International Committee on Taxonomy of Viruses (2020). Arch. Virol. 2020, 165, 2737–2748. [Google Scholar] [CrossRef]
- Reuter, G.; Boros, Á.; Delwart, E.; Pankovics, P. Novel seadornavirus (family Reoviridae) related to Banna virus in Europe. Arch. Virol. 2013, 158, 2163–2167. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; He, Y.; Zhou, Y.; Meng, J.; Zhu, W.; Chen, H.; Liao, D.; Man, Y. Isolation and genetic characterization of Mangshi virus: A newly discovered Seadornavirus of the Reoviridae family found in Yunnan Province, China. PLoS ONE 2015, 10, e0143601. [Google Scholar] [CrossRef]
- Attoui, H.; Mertens, P.P.C.; Becnel, J.; Belaganahalli, S.; Bergoin, M.; Brussaard, C.P.; Chappell, J.D.; Ciarlet, M.; del Vas, M.; Dermody, T.S.; et al. Family: Reoviridae. In Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q., Adams, M.J., Carstens, E.B., Lefkowitz, E.J., Eds.; Elsevier: Amsterdam, The Netherlands, 2012; pp. 541–637. [Google Scholar] [CrossRef]
- Tao, S.J.; Chen, B.Q. Studies of coltivirus in China. Chin. Med. J. 2005, 118, 581–586. [Google Scholar] [PubMed]
- Attoui, H.; Jaafar, F.M.; Belhouchet, M.; Tao, S.; Chen, B.; Liang, G.; Tesh, R.B.; de Micco, P.; de Lamballerie, X. Liao ning virus, a new Chinese seadornavirus that replicates in transformed and embryonic mammalian cells. J. Gen. Virol. 2006, 87, 199–208. [Google Scholar] [CrossRef]
- Ngoi, C.N.; Siqueira, J.; Li, L.; Deng, X.; Mugo, P.; Graham, S.M.; Price, M.A.; Sanders, E.J.; Delwart, E. The plasma virome of febrile adult Kenyans shows frequent parvovirus B19 infections and a novel arbovirus (Kadipiro virus). J. Gen. Virol. 2016, 97, 3359–3367. [Google Scholar] [CrossRef] [Green Version]
- Ngoi, C.N.; Siqueira, J.; Li, L.; Deng, X.; Mugo, P.; Graham, S.M.; Price, M.A. Corrigendum to: The plasma virome of febrile adult Kenyans shows frequent parvovirus B19 infections and a novel arbovirus (Kadipiro virus). J. Gen. Virol. 2017, 98, 517. [Google Scholar] [CrossRef] [Green Version]
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.W.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B. ICTV virus taxonomy profile: Polyomaviridae. J. Gen. Virol. 2017, 98, 1159–1160. [Google Scholar] [CrossRef]
- Cantalupo, P.G.; Buck, C.B.; Pipas, J.M. Complete genome sequence of a polyomavirus recovered from a pomona leaf-nosed bat (Hipposideros pomona) metagenome data set. Genome Announc. 2017, 5, e01053-16. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.; Fagbo, S.F.; Alagaili, A.N.; Nitido, A.; Williams, S.H.; Ng, J.; Lee, B.; Durosinlorun, A.; Garcia, J.A.; Jain, K.; et al. A viral metagenomic survey identifies known and novel mammalian viruses in bats from Saudi Arabia. PLoS ONE 2019, 14, e0214227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardmeier, I.S. Virome of Swiss Bats. Ph.D. Thesis, University of Zurich, Zurich, Switzerland, 2021. [Google Scholar] [CrossRef]
- Kemenesi, G.; Gellért, Á.; Dallos, B.; Görföl, T.; Boldogh, S.; Estók, P.; Marton, S.; Oldal, M.; Martella, V.; Bányai, K.; et al. Sequencing and molecular modeling identifies candidate members of Caliciviridae family in bats. Infect. Genet. Evol. 2016, 41, 227–232. [Google Scholar] [CrossRef]
- Yinda, C.K.; Zell, R.; Deboutte, W.; Zeller, M.; Conceição-Neto, N.; Heylen, E.; Maes, P.; Knowles, N.J.; Ghogomu, S.M.; Van Ranst, M.; et al. Highly diverse population of Picornaviridae and other members of the Picornavirales, in Cameroonian fruit bats. BMC Genom. 2017, 18, 249. [Google Scholar] [CrossRef] [Green Version]
- Debat, H.J. An RNA virome associated to the golden orb-weaver spider Nephila clavipes. Front. Microbiol. 2017, 8, 2097. [Google Scholar] [CrossRef] [Green Version]
- Oude Munnink, B.B.; Phan, M.V.T.; Simmonds, P.; Koopmans, M.P.G.; Kellam, P.; van der Hoek, L.; Cotten, M. Characterization of Posa and Posa-like virus genomes in fecal samples from humans, pigs, rats, and bats collected from a single location in Vietnam. Virus Evol. 2017, 3, vex022. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Vijayendran, D.; Chen, Y.; Bonning, B.C. Aphis glycines Virus 2, a novel insect virus with a unique genome structure. Viruses 2016, 8, 315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryabov, E.V.; Keane, G.; Naish, N.; Evered, C.; Winstanley, D. Densovirus induces winged morphs in asexual clones of the rosy apple aphid, Dysaphis plantaginea. Proc. Natl. Acad. Sci. USA 2009, 106, 8465–8470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.N.; Hsu, H.C.; Wang, S.W.; Lien, H.C.; Lu, H.T.; Peng, S.K. Entry of Scotophilus bat coronavirus-512 and severe acute respiratory syndrome coronavirus in human and multiple animal cells. Pathogens 2019, 8, 259. [Google Scholar] [CrossRef] [Green Version]
- De Wilde, A.H.; Snijder, E.J.; Kikkert, M.; van Hemert, M.J. Host factors in coronavirus replication. In Roles of Host Gene and Non-Coding RNA Expression in Virus Infection; Tripp, R.A., Tompkins, S.M., Eds.; Springer International Publishing: Cham, Switzerland, 2018; Volume 419, pp. 1–42. [Google Scholar] [CrossRef] [Green Version]
- Li, F. Structure, function, and evolution of coronavirus spike proteins. Annu. Rev. Virol. 2016, 3, 237–261. [Google Scholar] [CrossRef] [Green Version]
- Uddin, W.; Brändel, S.D.; Tschapka, M.; Page, R.; Rasche, A.; Corman, V.M.; Drosten, C.; Sommer, S. Astrovirus infections induce age-dependent dysbiosis in gut microbiomes of bats. ISME J. 2018, 12, 2883–2893. [Google Scholar] [CrossRef]
- Orłowska, A.; Smreczak, M.; Potyrało, P.; Bomba, A.; Trębas, P.; Rola, J. First detection of bat astroviruses (BtAstVs) among bats in Poland: The genetic BtAstVs diversity reveals multiple co-infection of bats with different strains. Viruses 2021, 13, 158. [Google Scholar] [CrossRef] [PubMed]
- Wohlgemuth, N.; Honce, R.; Schultz-Cherry, S. Infection, genetics and evolution astrovirus evolution and emergence. Infect. Genet. Evol. 2019, 69, 30–37. [Google Scholar] [CrossRef]
- Donato, C.; Vijaykrishna, D. The broad host range and genetic diversity of mammalian and avian astroviruses. Viruses 2017, 9, 102. [Google Scholar] [CrossRef] [Green Version]
- Barry, A.F.; Durães-Carvalho, R.; Oliveira-Filho, E.F.; Alfieri, A.A.; Van der Poel, W.H.M. High-Resolution phylogeny providing insights towards the epidemiology, zoonotic aspects and taxonomy of sapoviruses. Infect. Genet. Evol. 2017, 56, 8–13. [Google Scholar] [CrossRef]
- Gildow, F.E.; D’Arcy, C.J. Cytopathology and experimental host range of Rhopalosiphum padi virus, a small isometric RNA virus infecting cereal grain aphids. J. Invertebr. Pathol. 1990, 55, 245–257. [Google Scholar] [CrossRef]
- Bennett, A.J.; Bushmaker, T.; Cameron, K.; Ondzie, A.; Niama, F.R.; Parra, H.J.; Mombouli, J.V.; Olson, S.H.; Munster, V.J.; Goldberg, T.L. Diverse RNA viruses of arthropod origin in the blood of fruit bats suggest a link between bat and arthropod viromes. Virology 2019, 528, 64–72. [Google Scholar] [CrossRef]
- Attoui, H.; Charrel, R.N.; Billoir, F.; Cantaloube, J.F.; De Micco, P.; De Lamballerie, X. Comparative sequence analysis of American, European and Asian isolates of viruses in the genus Coltivirus. J. Gen. Virol. 1998, 79, 2481–2489. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Li, F.; Liu, A.; Lin, X.; Fu, S.; Song, J.; Liu, G.; Shao, N.; Tao, Z.; Wang, Q.; et al. Identification and genetic analysis of Kadipiro virus isolated in Shandong province, China. Virol. J. 2018, 15, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, C.; Kurth, A. European bats as carriers of viruses with zoonotic potential. Viruses 2014, 6, 3110–3128. [Google Scholar] [CrossRef] [Green Version]
- Seltmann, A.; Corman, V.M.; Rasche, A.; Drosten, C.; Czirják, G.; Bernard, H.; Struebig, M.J.; Voigt, C.C. Seasonal fluctuations of astrovirus, but not coronavirus shedding in bats inhabiting human-modified tropical forests. Ecohealth 2017, 14, 272–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kemenesi, G.; Dallos, B.; Gorfol, T.; Boldogh, S.; Estok, P.; Kurucz, K.; Kutas, A.; Foldes, F.; Oldal, M.; Nemeth, V.; et al. Molecular survey of RNA viruses in hungarian bats: Discovering novel astroviruses, coronaviruses, and caliciviruses. Vector Borne Zoonotic Dis. 2014, 14, 846–855. [Google Scholar] [CrossRef] [PubMed]
- Gloza-Rausch, F.; Ipsen, A.; Seebens, A.; Göttsche, M.; Panning, M.; Drexler, J.F.; Petersen, N.; Annan, A.; Grywna, K.; Müller, M.; et al. Detection and prevalence patterns of group I coronaviruses in bats, Northern Germany. Emerg. Infect. Dis. 2008, 14, 626–631. [Google Scholar] [CrossRef]
- Bentley, K.; Evans, D.J. Mechanisms and consequences of positive-strand RNA virus recombination. J. Gen. Virol. 2018, 99, 1345–1356. [Google Scholar] [CrossRef]
- Fagre, A.C.; Kading, R.C. Can bats serve as reservoirs for Arboviruses? Viruses 2019, 11, 215. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Sample ID | Species | Collection Time | Location | Previous Data | Virus Sequences Assembled in This Study |
|---|---|---|---|---|---|
| 13585-35 | M. dau | October 2014 | Mønsted | LR025670, LR025709 | BtCoV |
| 13585-58 | M. dau | October 2014 | Mønsted | LR025680, LR025712 | BtCoV, BtAstV, BtPV, RAAV |
| 21164-6 | M. dau | November 2015 | Mønsted | LR025732 | BtCoV, BtAstV, BtCV, BtPyV, KDV, BaV |
| OV-157 | M. dau | October 2018 | Mønsted | - | BtCoV, BtCV, RhPV |
| 18802-1 | M. das | November 2016 | Mønsted | LR025723 | BtCoV, RVH |
| B40-5 | P. pyg | August 2013 | Sollerup | LR025698, LR025733 | BtCoV |
| 7542-55 | P. pyg | June 2014 | Vadum | LR025676, LR025706 | BtCoV |
| NGS Dataset | D28-7 | D28-9 | D32-7 | D32-9 | D29-6 + D32-10 | D32-6 | D32-4 |
|---|---|---|---|---|---|---|---|
| Bat species | M. dau | M. dau | M. dau | M. dau | M. das | P. pyg | P. pyg |
| Sample no. | 13585-35 | 13585-58 | 21164-6 | OV-157 | 18802-1 | B40-5 | 7542-55 |
| Biosample ac. no. | SAMN 18653548 | SAMN 18653642 | SAMN 18661215 | SAMN 18680226 | SAMN 18665528 | SAMN 18672062 | SAMN 18672089 |
| Assigned reads (% of raw data) | 296,356 (23%) | 1,469,272 (42%) | 2,234,894 (63%) | 2,155,628 (62%) | 1,495,375 (49%) | 55,361 (19%) | 182,572 (50%) |
| Archaea | 2221 | 503 | 132 | 352 | 2078 | 207 | 360 |
| Bacteria | 189,602 | 1,121,102 | 28,900 | 164,552 | 1,447,060 | 39,417 | 148,103 |
| Eukaryota | 22,941 | 144,898 | 8868 | 18,813 | 18,251 | 2438 | 9956 |
| Viruses: | 72,440 | 189,014 | 2,195,308 | 1,961,910 | 8998 | 10,187 | 2871 |
| Unclassified vir. | 13,181 1 | 7865 1 | 3742 2 | 497,671 1 | 6 | 8 | 3 |
| Unclas. RNA vir. [43] | 44,631 3 | 40,180 3 | 6584 4 | 654,384 3 | 264 | 401 | 5 |
| Unclas. bacterial vir. | 26 | 2997 | - | 13 | 22 | 8 | 20 |
| Retro-transcribing vir. | 386 | 44 | 31 | 66 | 276 | 113 | 115 |
| dsDNA viruses | 1012 | 208 | 233 5 | 97 | 1565 | 84 | 188 |
| ssDNA viruses | - | 23 | - | - | 16 | 7 | - |
| dsRNA viruses | 458 6 | 29 | 2,113,874 7 | 770 8 | 90 9 | - | - |
| ssRNA viruses: | 9980 | 136,492 | 70,512 | 734,899 | 6708 | 9501 | 2517 |
| Unclassified vir. | 22 | 7242 10 | - | 306 | - | - | - |
| Coronavirinae | 6381 | 117,972 | 68,838 | 52,185 | 5934 | 7273 | 1173 |
| Astroviridae | 8 | 5754 | 744 | 2 | - | - | - |
| Tymovirales | 87 | 2811 | 54 | 15 | 7 | 191 | - |
| Nodaviridae | 8 | - | 357 | - | 6 | - | - |
| Picornavirales: | 3166 | 1094 | 435 13 | 681,091 | 41 | 1825 | 14 |
| Picornaviridae | - | 902 12 | - | 42 | - | 2 | - |
| Iflaviridae | 443 | 137 | - | 5063 15 | 12 | 1795 14 | - |
| Dicistroviridae | 2679 11 | 17 | 48 | 675,984 11 | 26 | 19 | 10 |
| Caliciviridae | - | - | 28 | 276 | - | - | - |
| Virus Description | Virus Strain Name | Mapped Reads | Average Coverage | Length nt. | Accession Number |
|---|---|---|---|---|---|
| Alphacoronavirus | BtCoV/13585-35/M.dau/DK/2014 | 8800 | 50 | 28,096 | MN535731 |
| BtCoV/13585-58/M.dau/DK/2014 | 140,000 | 1000 | 28,140 | MN535732 | |
| BtCoV/21164-6/M.dau/DK/2015 | 70,000 | 370 | 28,117 | MN543743 | |
| BtCoV/21164-6-alt/M.dau/DK/2015 | 20,000 | 120 | 28,092 | MZ218052 | |
| BtCoV/OV-157/M.dau/DK/2018 | 13,000 | 102 | 28,192 | MN535733 | |
| BtCoV/18802-1/M.das/DK/2016 | 9800 | 42 | 28,013 | MN535734 | |
| BtCoV/B40-5/P.pyg/DK/2013 | 9200 | 59 | 27,943 | MN482242 | |
| BtCoV/7542-55/P.pyg/DK/2014 | 1800 | 8 | 27,786 1 | MZ218060 | |
| Mamastrovirus | BtAstV/13585-58/M.dau/DK/2014 | 17,000 | 465 | 6557 | MN832787 |
| BtAstV/21164-6-A/M.dau/DK/2015 | 630 | 15 | 6619 | MZ218053 | |
| BtAstV/21164-6-B/M.dau/DK/2015 | 540 | 13 | 6558 2 | MZ218054 | |
| Calicivirus | BtCV/OV-157/M.dau/DK/2018 | 352 | 8 | 7486 3 | MZ218056 |
| BtCV/21164-6-A/M.dau/DK/2015 | 2 | 1.5 | 396 | MZ218057 | |
| BtCV/21164-6-B/M.dau/DK/2015 | 6 | 1.4 | 706 | MZ218058 | |
| Rotavirus H | RVH/18802-1/M.das/DK/2016 | 98 | 2.2 | 58–2169 4,5 | MZ218062- |
| MZ218070 | |||||
| Polyomavirus | BtPyV/21164-6/M.dau/DK/2015 | 710 | 17 | 4758 | MZ218055 |
| Picornavirus | BtPV/13585-58/M.dau/DK/2014 | 870 | 16 | 9300 | MZ218061 |
| Basavirus | BaV/21164-6/M.dau/DK/2015 | 8560 | 150 | 9155 | MW929926 |
| Kadipiro virus | KDV/21164-6/M.dau/DK/2015 | 1,960,000 | 3700–16,500 | 754–3750 6 | MN543741, |
| MN543742, | |||||
| MN543744- | |||||
| MN543753 | |||||
| Rhopalosiphum padi virus | RhPV/OV-157/M.dau/DK/2018 | 440,000 | 6490 | 10,009 | MN535735 |
| RhPV/OV-157-alt/M.dau/DK/2018 | 340,000 | 4600 | 10,010 | MZ218059 | |
| Rosy apple aphid virus | RAAV/13585-58/M.dau/DK/2014 | 5860 | 102 | 9742 | MW929927 |
| BtAstV Genomes | 1st BLASTn Hit | 1st BLASTn Hit Source | 2nd BLASTn Hit | 3rd BLASTn Hit |
|---|---|---|---|---|
| BtAstV/13585-58/M.dau/DK/2014 | KT894882 (93.7/379) | M. daubentonii, D15, September 2014, NRW | EU847161 (77.5/240) | EU847160 (77.5/240) |
| BtAstV/21164-6-A/M.dau/DK/2015 | KT894889 (98.2/381) | M. daubentonii, D13, September 2014, NRW | KT894883 (91.9/381) | KT894893 (92.0/375) |
| BtAstV/21164-6-B/M.dau/DK/2015 | KT894894 (99.5/381) | M. daubentonii, D10, July 2013, MV | KT894896 (99.2/381) | KT894895 (99.2/381) |
| RhPV/OV-157/M.dau/DK/2018 | RhPV/OV-157-alt/M.dau/DK/2018 | |||
|---|---|---|---|---|
| Nucleotide Comparison | (10,009 nt) | (10,010 nt) | ||
| Nt. Differences | Percent Identity | Nt. Differences | Percent Identity | |
| NC_001874 (10,011 nt) | 351 | 96.5 | 357 | 96.4 |
| - 5′-end (579 nt) | 47 | 91.9 | 48 | 91.7 |
| - ORF1 (5997 nt) | 230 | 96.2 | 231 | 96.2 |
| - IGR (533 nt) | 8 | 98.5 | 11 | 97.9 |
| - ORF2 (2451 nt) | 59 | 97.6 | 59 | 97.6 |
| - 3′-end (451 nt) | 7 | 98.4 | 8 | 98.2 |
| MF535298 (9966 nt) | 50 | 99.5 | 76 | 99.2 |
| - 5′-end (219 nt) | 0 | 100 | 0 | 100 |
| - ORF1 (5997 nt) | 19 | 99.7 | 48 | 99.2 |
| - IGR (533 nt) | 2 | 99.6 | 3 | 99.4 |
| - ORF2 (2451 nt) | 23 | 99.1 | 21 | 99.1 |
| - 3′-end (451 nt) | 6 | 98.7 | 4 | 99.1 |
| OV-157-alt (10,010 nt) | 106 | 98.9 | ||
| - 5′-end (579 and 580 nt) | 2 | 99.7 | ||
| - ORF1 (5997 nt) | 59 | 99.0 | ||
| - IGR (533 nt) | 5 | 99.1 | ||
| - ORF2 (2361 nt) | 31 | 98.7 | ||
| - 3′-end (539 nt) | 9 | 98.3 | ||
| Amino acid comparison: ORF1 | (1988 a.a.) | (1988 a.a.) | ||
| A.a. differences | Percent identity | A.a. differences | Percent identity | |
| NC_001874 (1988 a.a.) | 23 | 98.9 | 28 | 98.6 |
| MF535298 (1988 a.a.) | 3 | 99.9 | 4 | 99.8 |
| OV-157-alt (1988 a.a.) | 7 | 99.7 | ||
| Amino acid comparison: ORF2 | (788 a.a.) | (788 a.a.) | ||
| A.a. differences | Percent identity | A.a. differences | Percent identity | |
| NC_001874 (818 a.a.) | 11 | 98.6 | 13 | 98.4 |
| MF535298 (818 a.a.) | 6 | 99.2 | 6 | 99.2 |
| OV-157-alt (788 a.a.) | 4 | 99.5 | ||
| Genome Segment | Segment Length | Accession Number | 1st BLASTn Hit | 2nd BLASTn Hit | 3rd BLASTn Hit |
|---|---|---|---|---|---|
| Segment 1 | 3799 | MN543741 | KX884650 (86) | MG590148 (86) | AF133429 (84) |
| Segment 2 | 3016 | MN543742 | MG590140 (85) | KX884651 (84) | AF134509 (82) |
| Segment 3 | 2353 | MN543744 | MG590141 (84) | KX884652 (84) | AF134510 (81) |
| Segment 4 | 2121 | MN543745 | KX884653 (86) | AF134511 (84) | KX247778 (74) |
| Segment 5 | 1895 | MN543746 | MG590142 (83) | KX884654 (83) | AF134512 (81) |
| Segment 6 | 1659 | MN543747 | KX884655 (87) | AF134513 (86) | MG590143 (86) |
| Segment 7 | 1232 | MN543748 | MG590144 (92) | KX884656 (92) | AF052023 (90) |
| Segment 8 | 1083 | MN543749 | KX884657 (88) | AF052022 (85) | MG590145 (86) |
| Segment 9 | 1041 | MN543750 | KX884658 (91) | MG590150 (90) | AF052021 (88) |
| Segment 10 | 914 | MN543751 | KX884659 (87) | MG590151 (86) | AF052020 (84) |
| Segment 11 | 875 | MN543752 | KX884660 (85) | AF052019 (83) | MG590146 (83) |
| Segment 12 | 754 | MN543753 | MG590147 (93) | FJ159105 (90) | KX884661 (89) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lazov, C.M.; Belsham, G.J.; Bøtner, A.; Rasmussen, T.B. Full-Genome Sequences of Alphacoronaviruses and Astroviruses from Myotis and Pipistrelle Bats in Denmark. Viruses 2021, 13, 1073. https://doi.org/10.3390/v13061073
Lazov CM, Belsham GJ, Bøtner A, Rasmussen TB. Full-Genome Sequences of Alphacoronaviruses and Astroviruses from Myotis and Pipistrelle Bats in Denmark. Viruses. 2021; 13(6):1073. https://doi.org/10.3390/v13061073
Chicago/Turabian StyleLazov, Christina M., Graham J. Belsham, Anette Bøtner, and Thomas Bruun Rasmussen. 2021. "Full-Genome Sequences of Alphacoronaviruses and Astroviruses from Myotis and Pipistrelle Bats in Denmark" Viruses 13, no. 6: 1073. https://doi.org/10.3390/v13061073
APA StyleLazov, C. M., Belsham, G. J., Bøtner, A., & Rasmussen, T. B. (2021). Full-Genome Sequences of Alphacoronaviruses and Astroviruses from Myotis and Pipistrelle Bats in Denmark. Viruses, 13(6), 1073. https://doi.org/10.3390/v13061073