
Replication Kinetics of B.1.351 and B.1.1.7 SARS-CoV-2 Variants of Concern Including Assessment of a B.1.1.7 Mutant Carrying a Defective ORF7a Gene

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Patient Samples, Cell Culture and Virus Isolation
2.2. Next Generation Sequencing of Virus Isolates and Phylogenetic Analysis
2.3. Kinetic Replication Curve Experiments
2.4. Statistical Analysis
3. Results
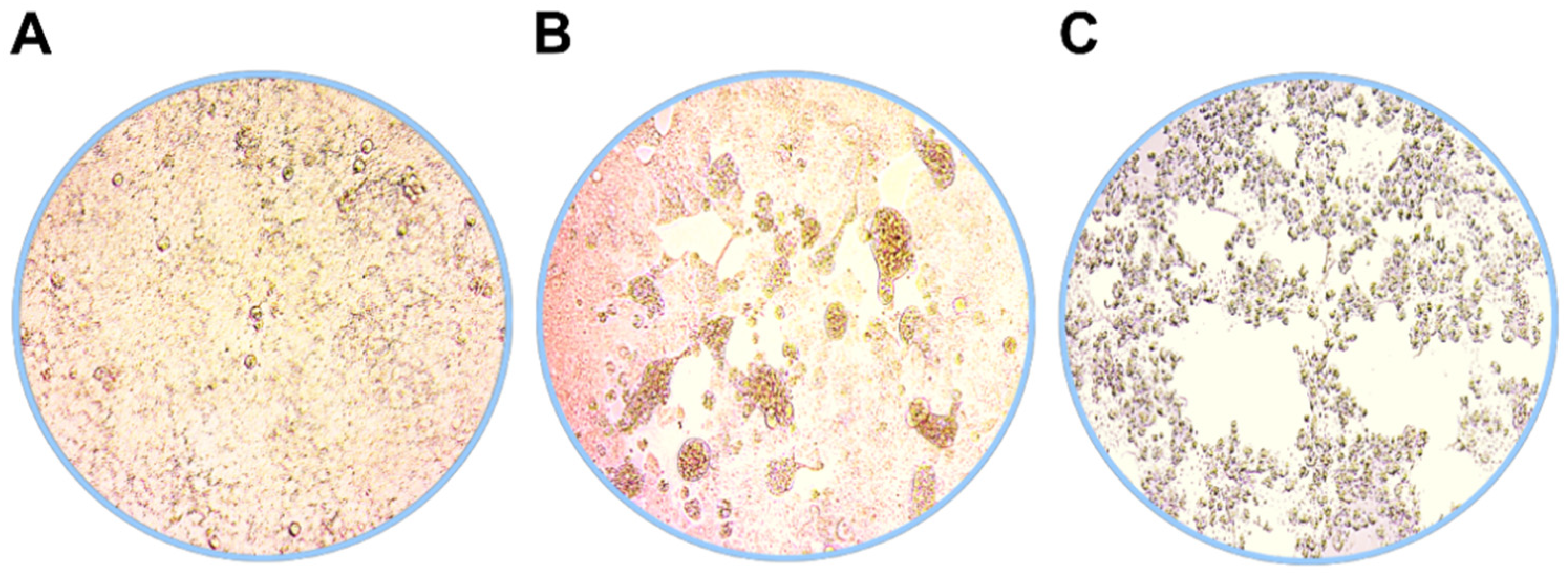
3.1. Virus Isolation
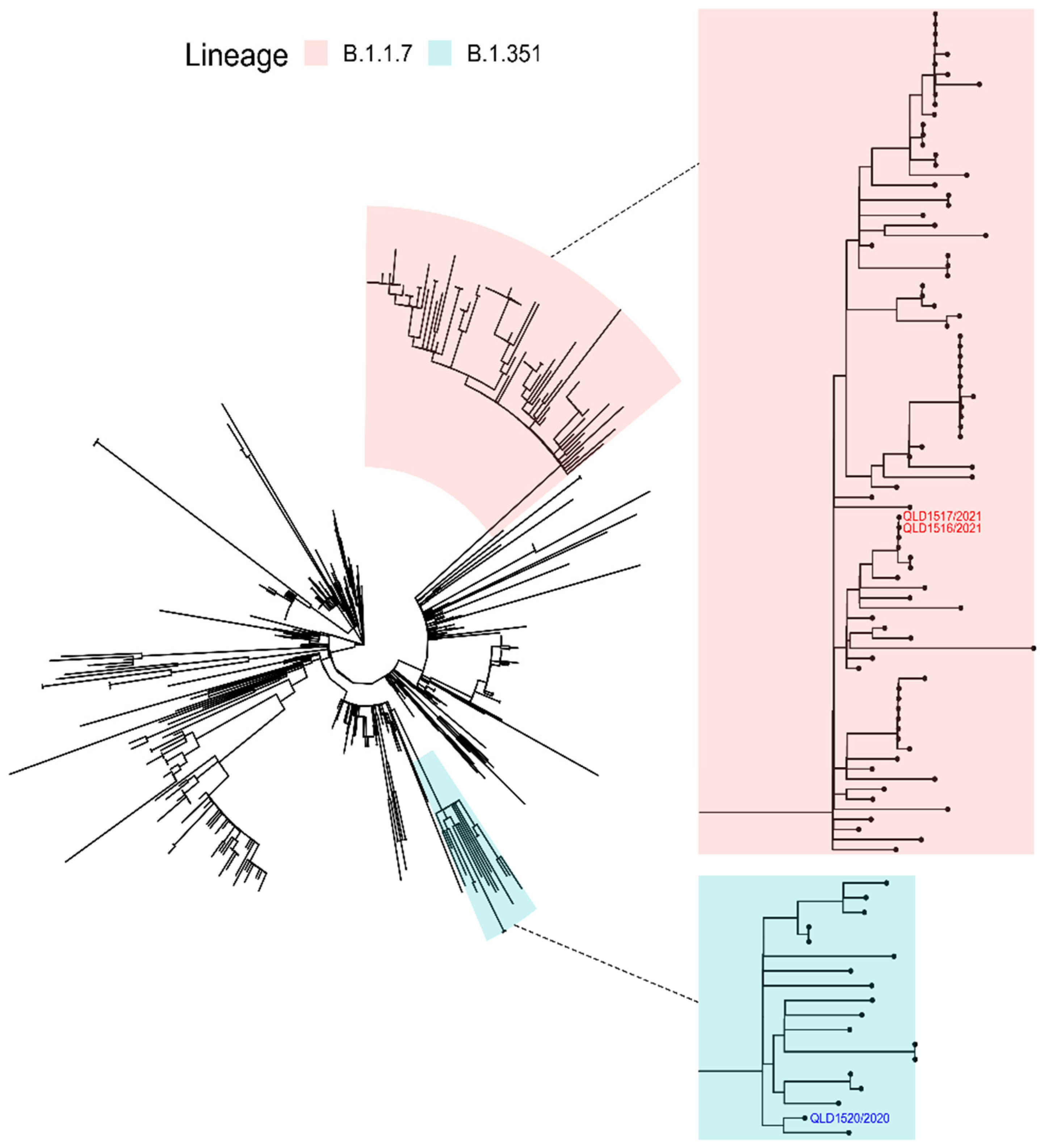
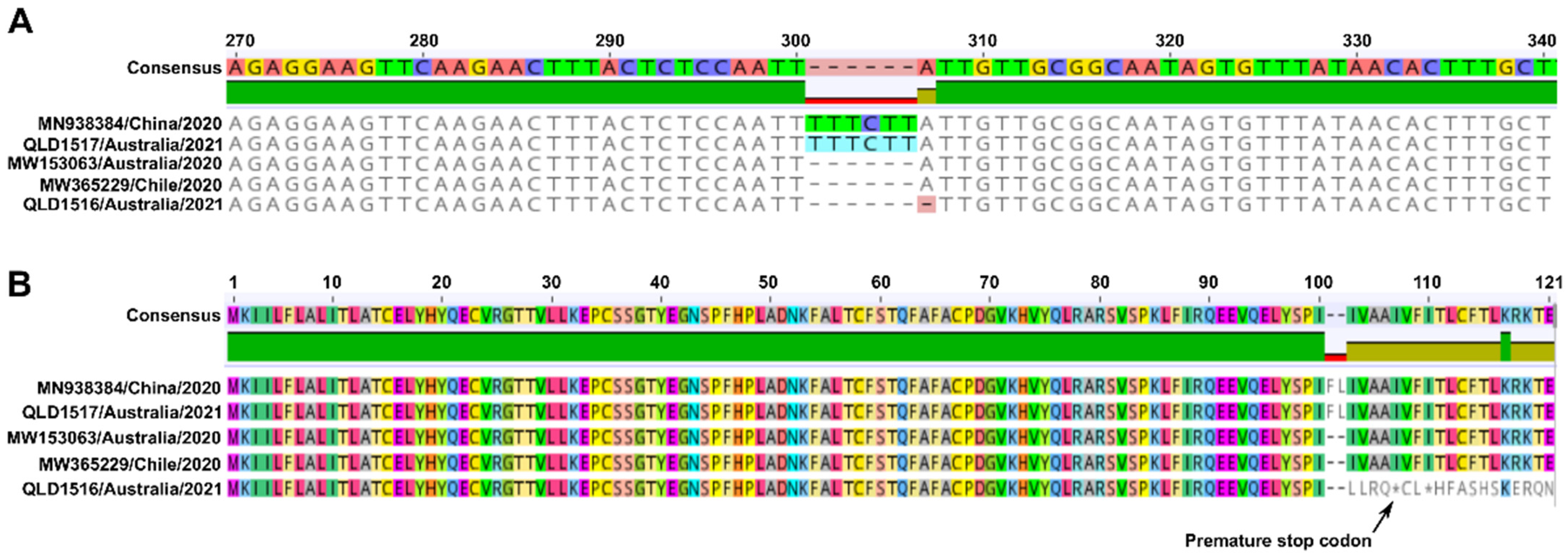
3.2. Next Generation Sequencing (NGS)
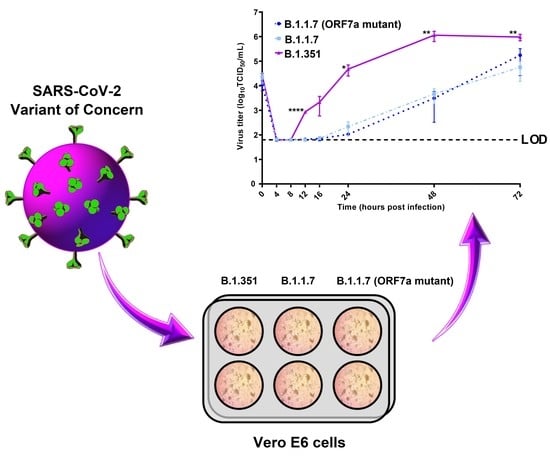
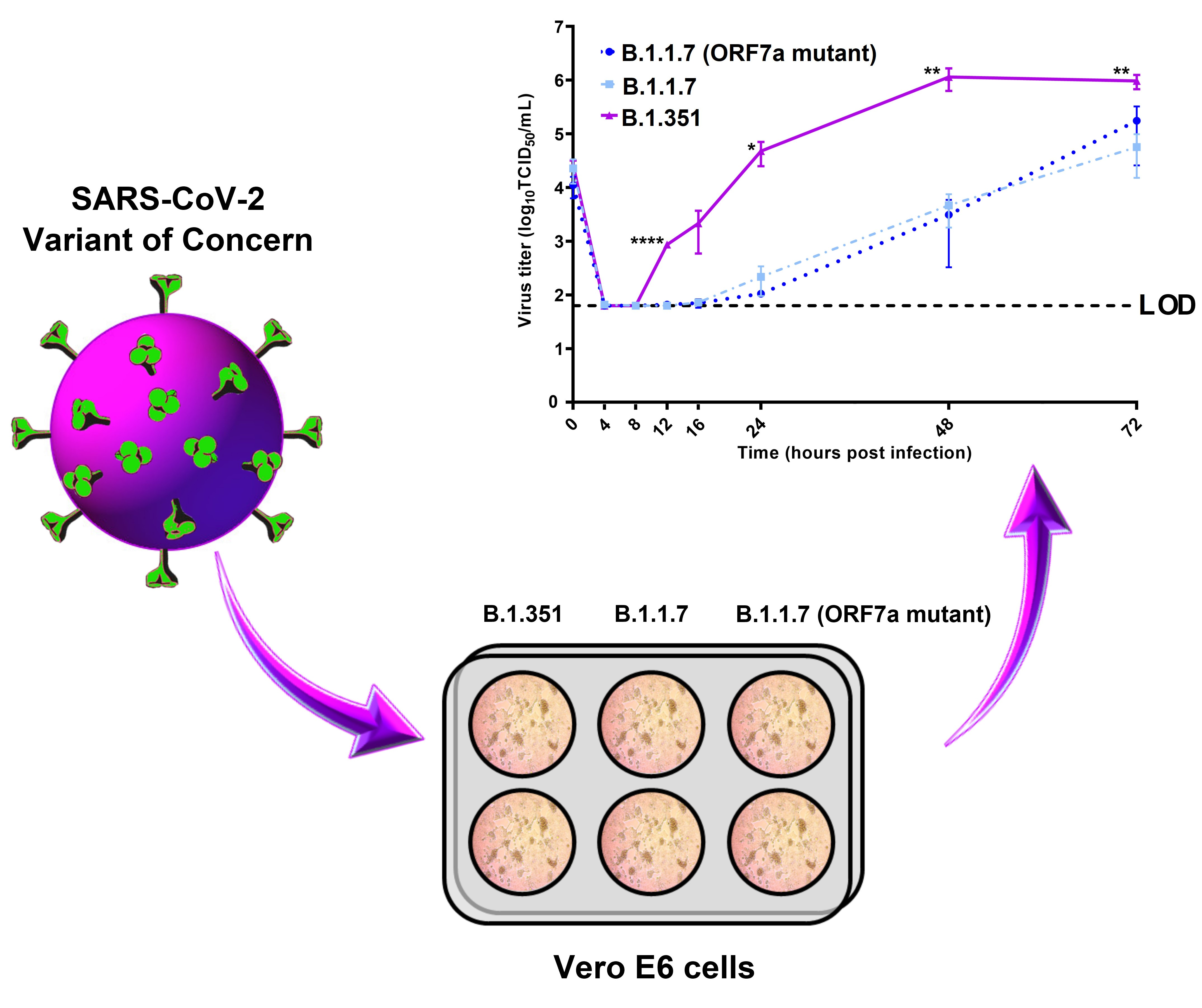
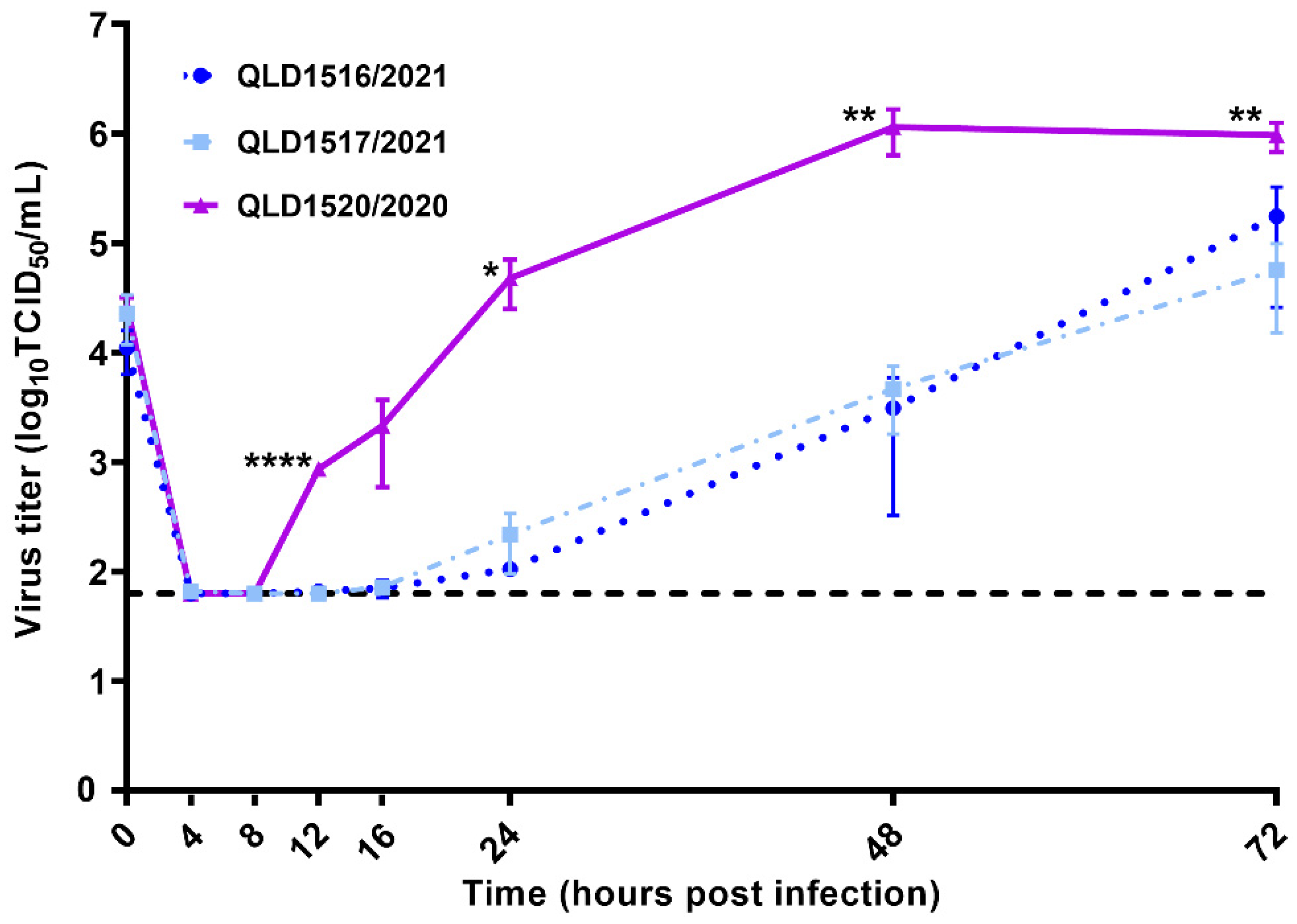
3.3. Kinetic Replication Curve Experiments
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chu, H.; Chan, J.F.-W.; Yuen, T.T.-T.; Shuai, H.; Yuan, S.; Wang, Y.; Hu, B.; Yip, C.C.-Y.; Tsang, J.O.-L.; Huang, X.; et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microbe 2020, 1, e14–e23. [Google Scholar] [CrossRef]
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. 2020. Available online: https://covid19.who.int/ (accessed on 3 March 2021).
- Harcourt, J.; Tamin, A.; Lu, X.; Kamili, S.; Sakthivel, S.K.; Murray, J.; Queen, K.; Tao, Y.; Paden, C.R.; Zhang, J.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with Coronavirus Disease, United States. Emerg. Infect. Dis. 2020, 26, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Conrol and Prevention. Science Brief: Emerging SARS-CoV-2 Variants. 2021. Available online: https://www.cdc.gov/coronavirus/2019-ncov/more/science-and-research/scientific-brief-emerging-variants.html (accessed on 18 March 2021).
- Plante, J.A.; Liu, Y.; Liu, J.; Xia, H.; Johnson, B.A.; Lokugamage, K.G.; Zhang, X.; Muruato, A.E.; Zou, J.; Fontes-Garfias, C.R.; et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature 2021, 592, 116–121. [Google Scholar] [CrossRef]
- Davies, N.G.; Jarvis, C.I.; Edmunds, W.J.; Jewell, N.P.; Diaz-Ordaz, K.; Keogh, R.H. Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nat. Cell Biol. 2021, 593, 270–274. [Google Scholar] [CrossRef]
- Khailany, R.A.; Safdar, M.; Ozaslan, M. Genomic characterization of a novel SARS-CoV-2. Gene Rep. 2020, 19, 100682. [Google Scholar] [CrossRef] [PubMed]
- Firth, A.E. A putative new SARS-CoV protein, 3c, encoded in an ORF overlapping ORF3a. J. Gen. Virol. 2020, 101, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Michel, C.J.; Mayer, C.; Poch, O.; Thompson, J.D. Characterization of accessory genes in coronavirus genomes. Virol. J. 2020, 17, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Pyke, A.; Mackay, I.; Moore, F.; Van, A.; Northill, J.; Finger, M.; Simpson, N.; Nair, N.; Burtonclay, P.; Moore, P.; et al. Culture of the Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2; f.2019-nCoV) v1. Available online: https://www.protocols.io/view/culture-of-the-severe-acute-respiratory-syndrome-c-bcduis6w (accessed on 11 March 2021).
- Pyke, A.T.; McMahon, J.; Burtonclay, P.; Nair, N.; De Jong, A. Genome Sequences of Chikungunya Virus Strains from Bangladesh and Thailand. Microbiol. Resour. Announc. 2020, 9, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Maio, N.; Walker, C.; Borges, R.; Weilguny, L.; Slodkowicz, G.; Goldman, N. Masking Strategies for SARS-CoV-2 Alignments. 2020. Available online: https://virological.org/t/masking-strategies-for-sars-cov-2-alignments/480 (accessed on 11 March 2021).
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, G. Using ggtree to visualize data on tree-like structures. Curr. Protoc. Bioinform. 2020, 69, e96. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; Du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Makoni, M. South Africa responds to new SARS-CoV-2 variant. Lancet 2021, 397, 267. [Google Scholar] [CrossRef]
- Roberts, M. South Africa Coronavirus Variant: What Is the Risk? BBC News, 2021. Available online: https://www.bbc.com/news/health-55534727(accessed on 28 April 2021).
- Steinhauser, G. South Africa Covid-19 Strain: What We Know about the New Variant. The Wall Street Journal, 2021. Available online: https://www.wsj.com/articles/the-new-covid-19-strain-in-south-africa-what-we-know-11609971229(accessed on 29 April 2021).
- Klein, B. UK Variant Is Now the Dominant Coronavirus Strain in the US, Says CDC Chief. Cable News Network, USA, 2021. Available online: https://edition.cnn.com/2021/04/07/us/uk-variant-dominant-coronavirus-strain/index.html(accessed on 28 April 2021).
- Mallapaty, S. What’s the risk of dying from a fast-spreading COVID-19 variant? Nat. Cell Biol. 2021, 590, 191–192. [Google Scholar] [CrossRef]
- Public Health England. SARS-CoV-2 Variants of Concern and Variants under Investigation in England, Technical Briefing 9. 2021. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/979818/Variants_of_Concern_VOC_Technical_Briefing_9_England.pdf (accessed on 27 April 2021).
- Luan, B.; Wang, H.; Huynh, T. Enhanced binding of the N501Y-mutated SARS-CoV-2 spike protein to the human ACE2 receptor: Insights from molecular dynamics simulations. FEBS Lett. 2021, 595, 1454–1461. [Google Scholar] [CrossRef] [PubMed]
- Nemudryi, A.; Nemudraia, A.; Wiegand, T.; Nichols, J.; Snyder, D.T.; Hedges, J.F.; Cicha, C.; Lee, H.; Vanderwood, K.K.; Bimczok, D.; et al. SARS-CoV-2 genomic surveillance identifies naturally occurring truncations of ORF7a that limit immune suppression. medRxiv 2021. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pyke, A.T.; Nair, N.; van den Hurk, A.F.; Burtonclay, P.; Nguyen, S.; Barcelon, J.; Kistler, C.; Schlebusch, S.; McMahon, J.; Moore, F. Replication Kinetics of B.1.351 and B.1.1.7 SARS-CoV-2 Variants of Concern Including Assessment of a B.1.1.7 Mutant Carrying a Defective ORF7a Gene. Viruses 2021, 13, 1087. https://doi.org/10.3390/v13061087
Pyke AT, Nair N, van den Hurk AF, Burtonclay P, Nguyen S, Barcelon J, Kistler C, Schlebusch S, McMahon J, Moore F. Replication Kinetics of B.1.351 and B.1.1.7 SARS-CoV-2 Variants of Concern Including Assessment of a B.1.1.7 Mutant Carrying a Defective ORF7a Gene. Viruses. 2021; 13(6):1087. https://doi.org/10.3390/v13061087
Chicago/Turabian StylePyke, Alyssa T., Neelima Nair, Andrew F. van den Hurk, Peter Burtonclay, Son Nguyen, Jean Barcelon, Carol Kistler, Sanmarié Schlebusch, Jamie McMahon, and Frederick Moore. 2021. "Replication Kinetics of B.1.351 and B.1.1.7 SARS-CoV-2 Variants of Concern Including Assessment of a B.1.1.7 Mutant Carrying a Defective ORF7a Gene" Viruses 13, no. 6: 1087. https://doi.org/10.3390/v13061087
APA StylePyke, A. T., Nair, N., van den Hurk, A. F., Burtonclay, P., Nguyen, S., Barcelon, J., Kistler, C., Schlebusch, S., McMahon, J., & Moore, F. (2021). Replication Kinetics of B.1.351 and B.1.1.7 SARS-CoV-2 Variants of Concern Including Assessment of a B.1.1.7 Mutant Carrying a Defective ORF7a Gene. Viruses, 13(6), 1087. https://doi.org/10.3390/v13061087