
Field and Molecular Epidemiology: How Viral Sequencing Changed Transmission Inferences in the First Portuguese SARS-CoV-2 Infection Cluster

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Number of Individuals and RT-PCR Test
2.2. Viral Whole-Genome-Sequencing
2.3. Serological Tests
3. Results
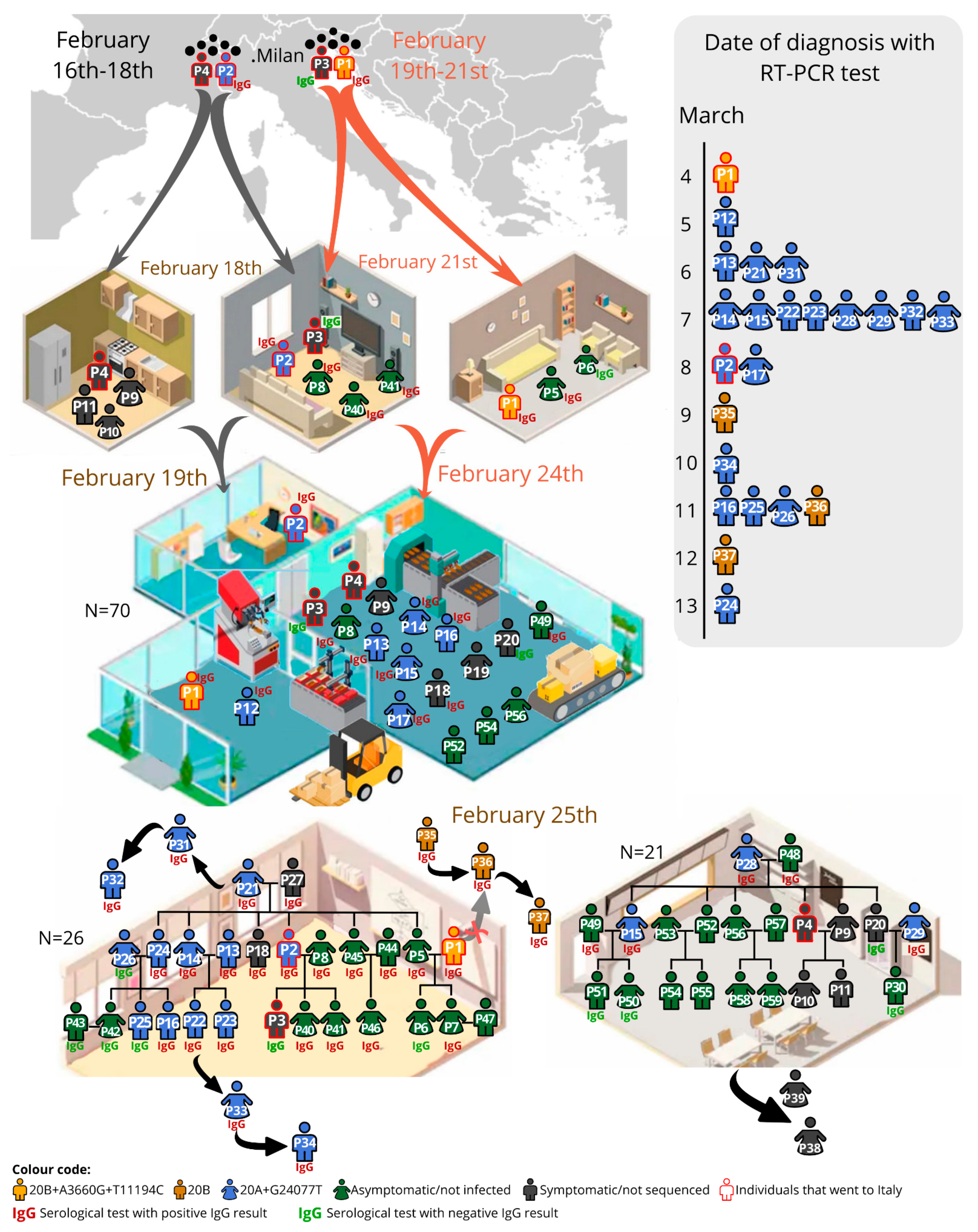
3.1. Case Study
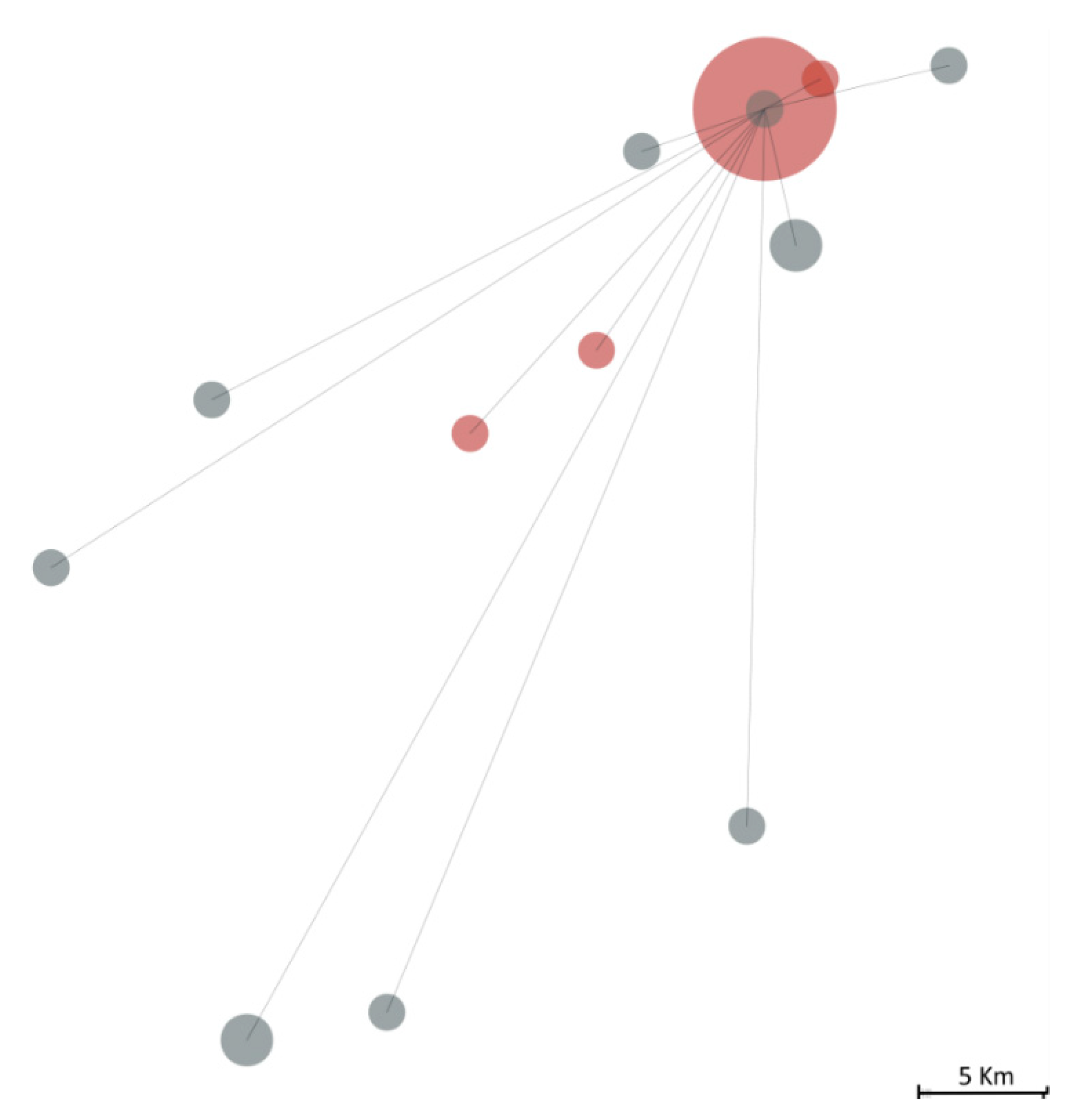
3.2. Description of the Transmission Chain
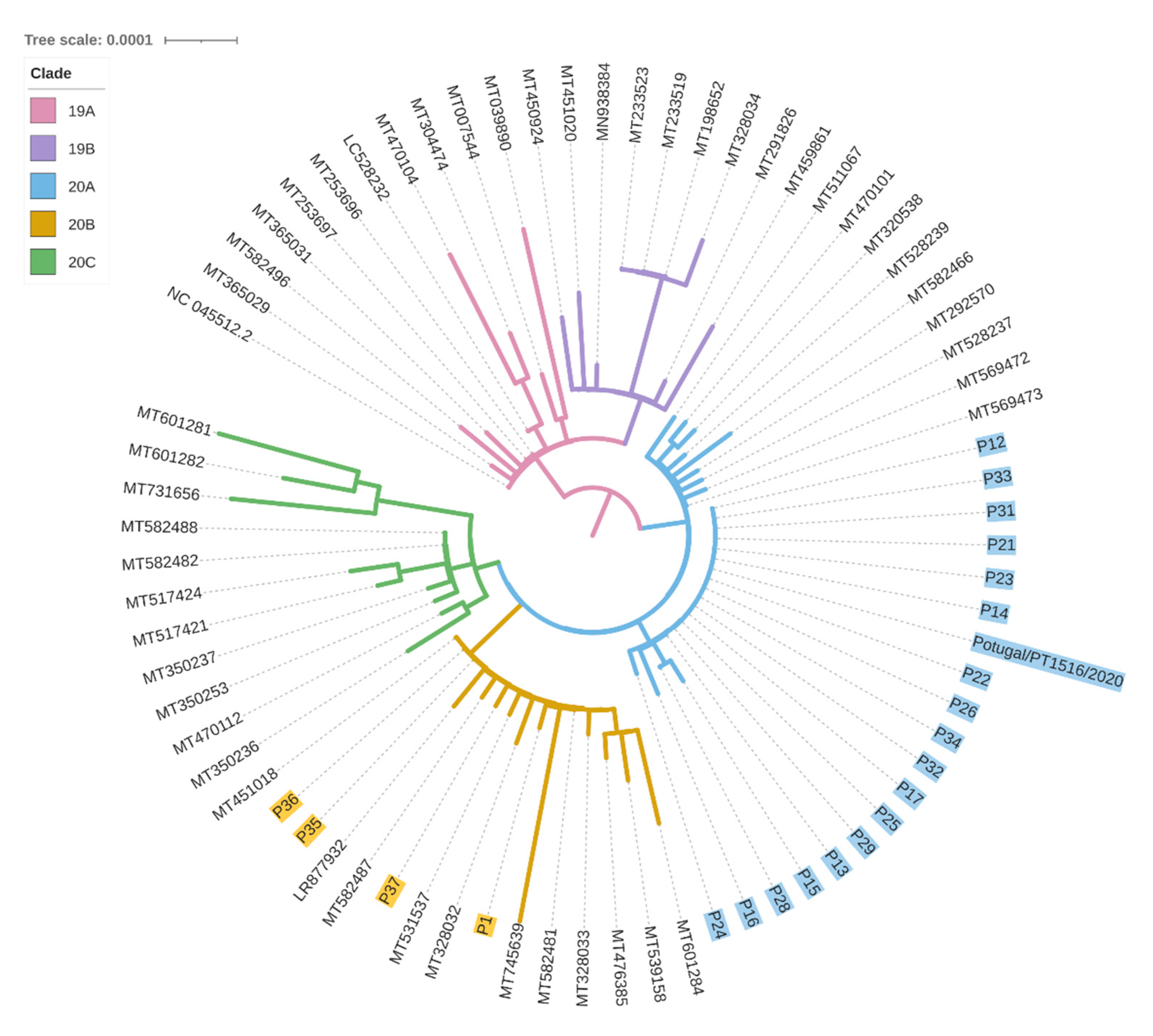
3.3. Viral Sequencing Interpretation
3.4. Viral Sequencing Contextualisation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adam, D.C.; Wu, P.; Wong, J.Y.; Lau, E.H.; Tsang, T.K.; Cauchemez, S.; Leung, G.M.; Cowling, B.J. Clustering and superspreading potential of SARS-CoV-2 infections in Hong Kong. Nat. Med. 2020, 26, 1714–1719. [Google Scholar] [CrossRef]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef]
- Majra, D.; Benson, J.; Pitts, J.; Stebbing, J. SARS-CoV-2 (COVID-19) superspreader events. J. Infect. 2021, 82, 36–40. [Google Scholar] [CrossRef]
- Gómez-Carballa, A.; Bello, X.; Pardo-Seco, J.; Martinón-Torres, F.; Salas, A. Mapping genome variation of SARS-CoV-2 worldwide highlights the impact of COVID-19 super-spreaders. Genome Res. 2020, 30, 1434–1448. [Google Scholar] [CrossRef]
- Alteri, C.; Cento, V.; Piralla, A.; Costabile, V.; Tallarita, M.; Colagrossi, L.; Renica, S.; Giardina, F.; Novazzi, F.; Gaiarsa, S.; et al. Genomic epidemiology of SARS-CoV-2 reveals multiple lineages and early spread of SARS-CoV-2 infections in Lombardy, Italy. Nat. Commun. 2021, 12, 434. [Google Scholar] [CrossRef] [PubMed]
- Borges, V.; Isidro, J.; Cortes-Martins, H.; Duarte, S.; Vieira, L.; Leite, R.; Gordo, I.; Caetano, C.P.; Nunes, B.; Sá, R.; et al. Massive dissemination of a SARS-CoV-2 Spike Y839 variant in Portugal. Emerg. Microbes Infect. 2020, 9, 2488–2496. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell 2020, 182, 812–827.e19. [Google Scholar] [CrossRef]
- Wise, J. Covid-19: New coronavirus variant is identified in UK. BMJ 2020, 371, m4857. [Google Scholar] [CrossRef]
- Cherian, S.; Potdar, V.; Jadhav, S.; Yadav, P.; Gupta, N.; Das, M.; Rakshit, P.; Singh, S.; Abraham, P.; Panda, S.; et al. Convergent evolution of SARS-CoV-2 spike mutations, L452R, E484Q and P681R, in the second wave of COVID-19 in Maharashtra, India. bioRxiv 2021. [Google Scholar] [CrossRef]
- Rasmussen, S.A.; Goodman, R.A. The CDC Field Epidemiology Manual; Oxford University Press: New York, NY, USA, 2019. [Google Scholar]
- WHO. Genomic Sequencing of SARS-CoV-2: A Guide to Implementation for Maximum Impact on Public Health; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Taylor, J.; Carter, R.J.; Lehnertz, N.; Kazazian, L.; Sullivan, M.; Wang, X.; Garfin, J.; Diekman, S.; Plumb, M.; Bennet, M.E.; et al. Serial Testing for SARS-CoV-2 and Virus Whole Genome Sequencing Inform Infection Risk at Two Skilled Nursing Facilities with COVID-19 Outbreaks, Minnesota, April–June 2020. Morb. Mortal. Weekly Rep. 2020, 69, 1288–1295. [Google Scholar] [CrossRef]
- Karmarkar, E.N.; Blanco, I.; Amornkul, P.N.; DuBois, A.; Deng, X.; Moonan, P.K.; Rubenstein, B.L.; Miller, D.A.; Kennedy, I.; Yu, J.; et al. Timely Intervention and Control of a Novel Coronavirus (COVID-19) Outbreak at a Large Skilled Nursing Facility, San Francisco, CA, USA, 2020. Infect. Control. Hosp. Epidemiol. 2020, 1–20. [Google Scholar] [CrossRef]
- Safdar, N.; Moreno, G.K.; Braun, K.M.; Friedrich, T.C.; O’Connor, D.H. Using Virus Sequencing to Determine Source of SARS-CoV-2 Transmission for Healthcare Worker. Emerg. Infect. Dis. 2020, 26, 2489–2491. [Google Scholar] [CrossRef] [PubMed]
- Wallace, M.; James, A.E.; Silver, R.; Koh, M.; Tobolowsky, F.A.; Simonson, S.; Gold, J.A.W.; Fukunaga, R.; Njuguna, H.; Bordelon, K.; et al. Rapid Transmission of Severe Acute Respiratory Syndrome Coronavirus 2 in Detention Facility, Louisiana, USA, May-June 2020. Emerg. Infect. Dis. 2021, 27, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Swadi, T.; Geoghegan, J.L.; Devine, T.; McElnay, C.; Sherwood, J.; Shoemack, P.; Ren, X.; Storey, M.; Jefferies, S.; Smit, E.; et al. Genomic Evidence of In-Flight Transmission of SARS-CoV-2 Despite Predeparture Testing. Emerg. Infect. Dis. 2021, 27. [Google Scholar] [CrossRef] [PubMed]
- Khanh, N.C.; Thai, P.Q.; Quach, H.L.; Thi, N.A.H.; Dinh, P.C.; Duong, T.N.; Mai, L.T.Q.; Nghia, N.D.; Tu, T.A.; Quang, L.N.; et al. Transmission of SARS-CoV 2 During Long-Haul Flight. Emerg. Infect. Dis. 2020, 26, 2617–2624. [Google Scholar] [CrossRef] [PubMed]
- Chau, N.V.V.; Hong, N.T.T.; Ngoc, N.M.; Thanh, T.T.; Khanh, P.N.Q.; Nguyet, L.A.; Nhu, L.N.T.; Ny, N.T.H.; Man, D.N.H.; Hang, V.T.T.; et al. Superspreading Event of SARS-CoV-2 Infection at a Bar, Ho Chi Minh City, Vietnam. Emerg. Infect. Dis. 2021, 27, 310–314. [Google Scholar] [CrossRef] [PubMed]
- Pedro, N.; Silva, C.N.; Magalhaes, A.C.; Cavadas, B.; Rocha, A.M.; Moreira, A.C.; Gomes, S.; Silva, D.; Sobrinho-Simoes, J.; Ramos, A.; et al. Dynamics of a dual SARS-CoV-2 lineage co-infection on a prolonged viral shedding COVID-19 case: Insights into clinical severity and disease duration. Microorganisms 2021, 9, 300. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Pickett, B.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef] [Green Version]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree OF Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256–W259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDade, T.W.; McNally, E.M.; Zelikovich, A.S.; D’Aquila, R.; Mustanski, B.; Miller, A.; Vaught, L.A.; Reiser, N.L.; Bogdanovic, E.; Fallon, K.S.; et al. High seroprevalence for SARS-CoV-2 among household members of essential workers detected using a dried blood spot assay. PLoS ONE 2020, 15, e0237833. [Google Scholar] [CrossRef] [PubMed]
- Gallais, F.; Velay, A.; Nazon, C.; Wendling, M.J.; Partisani, M.; Sibilia, J.; Candon, S.; Fafi-Kremer, S. Intrafamilial Exposure to SARS-CoV-2 Associated with Cellular Immune Response without Seroconversion, France. Emerg. Infect. Dis. 2021, 27, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Schwarzkopf, S.; Krawczyk, A.; Knop, D.; Klump, H.; Heinold, A.; Heinemann, F.M.; Thümmler, L.; Temme, C.; Breyer, M.; Witzke, O.; et al. Cellular Immunity in COVID-19 Convalescents with PCR-Confirmed Infection but with Undetectable SARS-CoV-2-Specific IgG. Emerg. Infect. Dis. 2021, 27, 122. [Google Scholar] [CrossRef]
- Li, W.; Su, Y.Y.; Zhi, S.S.; Huang, J.; Zhuang, C.L.; Bai, W.Z.; Wan, Y.; Meng, X.R.; Zhang, L.; Zhou, Y.B.; et al. Virus shedding dynamics in asymptomatic and mildly symptomatic patients infected with SARS-CoV-2. Clin. Microbiol. Infect. 2020, 26, 1556.e1–1556.e6. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ID | Transmission Event | Transmission Level | Viral Sequence | |||||
|---|---|---|---|---|---|---|---|---|
| Factory | Family1 | Family2 | Clade | Other Non-Clade Basal Mutations (%) | ||||
| P1 | X | X | 0 | 20B | A3660G (93%) | T11194C (55%) | ||
| P35 | 0 | 20B | ||||||
| P36 | 1 (P35) | 20B | ||||||
| P37 | 2 (P36) | 20B | G4432T (99%) | |||||
| P2 * | X | X | 0 | 20A | G24077T | |||
| P12 | X | 1 (P2) | 20A | G24077T (100%) | ||||
| P13 | X | X | 1 (P2) | 20A | G24077T (100%) | |||
| P14 | X | X | 1 (P2) | 20A | G24077T (100%) | |||
| P15 | X | X | 1 (P2) | 20A | G24077T (100%) | T14418C (54%) | G26634T (100%) | |
| P16 | X | X | 1 (P2) | 20A | G24077T (100%) | C2062T (70%) | C6279A (65%) | |
| P17 | X | 1 (P2) | 20A | G24077T (86%) | ||||
| P21 | X | 1 (P2) | 20A | G24077T (100%) | ||||
| P22 | X | 1 (P2) or 2 (P13/P14) | 20A | G24077T (100%) | ||||
| P23 | X | 1 (P2) or 2 (P13/P14) | 20A | G24077T (100%) | ||||
| P24 | X | 1 (P2) | 20A | G24077T (100%) | C4456T (82%) | |||
| P25 | X | 1 (P2) | 20A | G24077T (98%) | ||||
| P26 | X | 1 (P2) | 20A | G24077T (100%) | ||||
| P28 | X | 2 (P15) | 20A | G24077T (100%) | G26634T (73%) | |||
| P29 | X | 2 (P4/P20) | 20A | G24077T (100%) | ||||
| P31 | 2 (P21) | 20A | G24077T (100%) | |||||
| P32 | 3 (P31) | 20A | G24077T (100%) | |||||
| P33 | 2 or 3 (P22) | 20A | G24077T (100%) | |||||
| P34 | 3 or 4 (P33) | 20A | G24077T (100%) | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pedro, N.; Fernandes, V.; Cavadas, B.; Guimarães, J.T.; Barros, H.; Tavares, M.; Pereira, L. Field and Molecular Epidemiology: How Viral Sequencing Changed Transmission Inferences in the First Portuguese SARS-CoV-2 Infection Cluster. Viruses 2021, 13, 1116. https://doi.org/10.3390/v13061116
Pedro N, Fernandes V, Cavadas B, Guimarães JT, Barros H, Tavares M, Pereira L. Field and Molecular Epidemiology: How Viral Sequencing Changed Transmission Inferences in the First Portuguese SARS-CoV-2 Infection Cluster. Viruses. 2021; 13(6):1116. https://doi.org/10.3390/v13061116
Chicago/Turabian StylePedro, Nicole, Veronica Fernandes, Bruno Cavadas, João Tiago Guimarães, Henrique Barros, Margarida Tavares, and Luisa Pereira. 2021. "Field and Molecular Epidemiology: How Viral Sequencing Changed Transmission Inferences in the First Portuguese SARS-CoV-2 Infection Cluster" Viruses 13, no. 6: 1116. https://doi.org/10.3390/v13061116
APA StylePedro, N., Fernandes, V., Cavadas, B., Guimarães, J. T., Barros, H., Tavares, M., & Pereira, L. (2021). Field and Molecular Epidemiology: How Viral Sequencing Changed Transmission Inferences in the First Portuguese SARS-CoV-2 Infection Cluster. Viruses, 13(6), 1116. https://doi.org/10.3390/v13061116