
Oncolytic Virotherapy Treatment of Breast Cancer: Barriers and Recent Advances


Abstract
:1. Introduction
2. Preclinical Evidence of OV in Breast Cancer
3. Barriers to OV
4. Combination with Chemotherapy
5. Combination with Radiotherapy
6. Combination with Immunotherapies
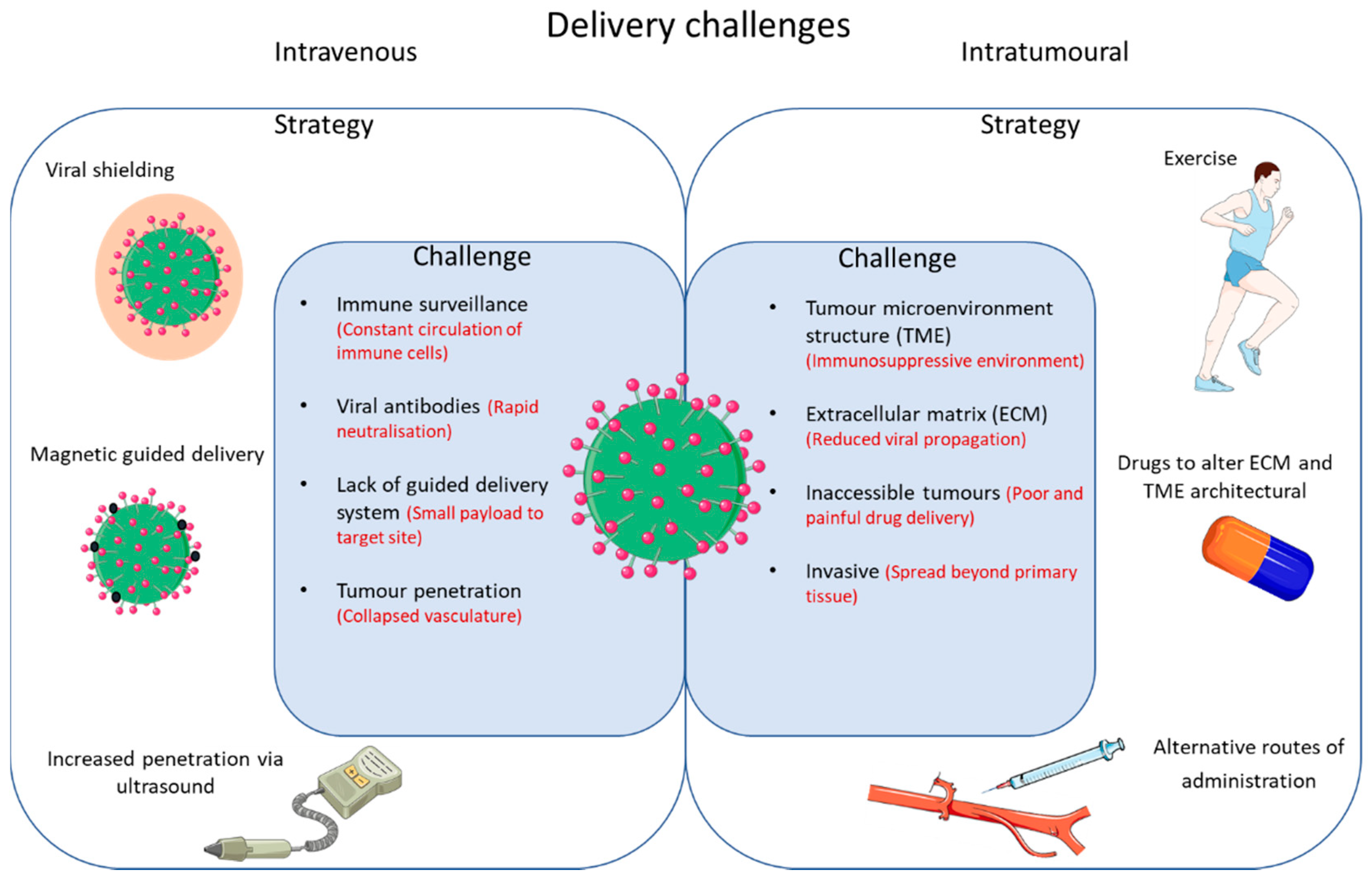
7. Overcoming Barriers of Intravenous Delivery OV
8. Overcoming Barriers Using a Targeted Delivery Approach—Magnetic Guided Delivery
9. Overcoming Breast Cancer-Specific Challenges
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- UK Cancer Research. Breast Cancer Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/breast-cancer (accessed on 22 March 2021).
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sørlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Carrick, S.; Parker, S.; Thornton, C.E.; Ghersi, D.; Simes, J.; Wilcken, N. Single agent versus combination chemotherapy for metastatic breast cancer. Cochrane Database Syst. Rev. 2009, 2009, CD003372. [Google Scholar] [CrossRef] [PubMed]
- Kelly, E.; Russell, S.J. History of oncolytic viruses: Genesis to genetic engineering. Mol. Ther. 2007, 15, 651–659. [Google Scholar] [CrossRef]
- Puzanov, I.; Milhem, M.M.; Minor, D.; Hamid, O.; Li, A.; Chen, L.; Chastain, M.; Gorski, K.; Anderson, A.; Chou, J.; et al. Talimogene laherparepvec in combination with ipilimumab in previously untreated, unresectable stage IIIB-IV melanoma. J. Clin. Oncol. 2016, 34, 2619–2626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Funchain, P.; Song, J.M.; Rayman, P.; Tannenbaum, C.; Ko, J.; Mcnamara, M.; Diaz-Montero, C.M.; Gastman, B. Talimogene Laherparepvec combined with anti-PD-1 based immunotherapy for unresectable stage III-IV melanoma: A case series. J. Immunother. Cancer 2018, 6, 1–7. [Google Scholar] [CrossRef]
- Wein, L.; Luen, S.J.; Savas, P.; Salgado, R.; Loi, S. Checkpoint blockade in the treatment of breast cancer: Current status and future directions. Br. J. Cancer 2018, 119, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Martini, V.; D’Avanzo, F.; Maggiora, P.M.; Varughese, F.M.; Sica, A.; Gennari, A. Oncolytic virotherapy: New weapon for breast cancer treatment. Ecancermedicalscience 2020, 14, 1149. [Google Scholar] [CrossRef]
- Oliva, S.; Gambella, M.; Boccadoro, M.; Bringhen, S. Systemic virotherapy for multiple myeloma. Expert Opin. Biol. Ther. 2017, 17, 1375–1387. [Google Scholar] [CrossRef]
- Ito, N.; Demarco, R.A.; Mailliard, R.B.; Han, J.; Rabinowich, H.; Kalinski, P.; Stolz, N.B.; Zeh, H.J.; Lotze, M.T. Cytolytic cells induce HMGB1 release from melanoma cell lines. J. Leukoc. Biol. 2006, 81, 75–83. [Google Scholar] [CrossRef]
- Bourgeois-Daigneault, M.-C.; St-Germain, L.; Roy, D.G.; Pelin, A.; Aitken, A.S.; Arulanandam, R.; Falls, T.; Garcia, V.; Diallo, J.-S.; Bell, J.C. Combination of Paclitaxel and MG1 oncolytic virus as a successful strategy for breast cancer treatment. Breast Cancer Res. 2016, 18, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Bramante, S.; Koski, A.; Liikanen, I.; Vassilev, L.; Oksanen, M.; Siurala, M.; Heiskanen, R.; Hakonen, T.; Joensuu, T.; Kanerva, A.; et al. Oncolytic virotherapy for treatment of breast cancer, including triple-negative breast cancer. OncoImmunology 2015, 5, e1078057. [Google Scholar] [CrossRef] [Green Version]
- Gholami, S.; Chen, C.-H.; Gao, S.; Lou, E.; Fujisawa, S.; Carson, J.; E Nnoli, J.; Chou, T.-C.; Bromberg, J.; Fong, Y. Role of MAPK in oncolytic herpes viral therapy in triple-negative breast cancer. Cancer Gene Ther. 2014, 21, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Gholami, S.; Marano, A.; Chen, N.G.; Aguilar, R.J.; Frentzen, A.; Chen, C.-H.; Lou, E.; Fujisawa, S.; Eveno, C.; Belin, L.; et al. A novel vaccinia virus with dual oncolytic and anti-angiogenic therapeutic effects against triple-negative breast cancer. Breast Cancer Res. Treat. 2014, 148, 489–499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gil, M.; Seshadri, M.; Komorowski, M.P.; Abrams, S.I.; Kozbor, D. Targeting CXCL12/CXCR4 signaling with oncolytic virotherapy disrupts tumor vasculature and inhibits breast cancer metastases. Proc. Natl. Acad. Sci. USA 2013, 110, E1291–E1300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.C.C.; Coffin, R.S.; Davis, C.J.; Graham, N.J.; Groves, N.; Guest, P.J.; Harrington, K.J.; James, N.D.; Love, C.A.; McNeish, I.; et al. A phase I study of OncoVEXGM-CSF, a second-generation oncolytic herpes simplex virus expressing granulocyte macrophage colony-stimulating factor. Clin. Cancer Res. 2006, 12, 6737–6747. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zeng, W.; Huang, Y.; Zhang, Q.; Hu, P.; Rabkin, S.D.; Liu, R. Treatment of breast cancer stem cells with oncolytic herpes simplex virus. Cancer Gene Ther. 2012, 19, 707–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- OBryan, S.M.; Mathis, J.M. Oncolytic virotherapy for breast cancer treatment. Curr. Gene Ther. 2018, 18, 192–205. [Google Scholar] [CrossRef]
- Chaurasiya, S.; Fong, Y. Viroimmunotherapy for breast cancer: Promises, problems and future directions. Cancer Gene Ther. 2020, 1–12. [Google Scholar] [CrossRef]
- Koski, A.; Kangasniemi, L.; Escutenaire, S.; Pesonen, S.; Cerullo, V.; Diaconu, I.; Nokisalmi, P.; Raki, M.; Rajecki, M.; Guse, K.; et al. Treatment of cancer patients with a serotype 5/3 chimeric oncolytic adenovirus expressing GMCSF. Mol Ther. 2010, 18, 1874–1884. [Google Scholar] [CrossRef]
- Li, W.; Li, Y.; Cui, Y.; Li, S.; Zhu, Y.; Shang, C.; Song, G.; Liu, Z.; Xiu, Z.; Cong, J.; et al. Anti-tumour effects of a dual cancer-specific oncolytic adenovirus on Breast Cancer Stem cells. J. Cell. Mol. Med. 2021, 25, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Jiang, H.; Cheng, L.; Liu, R. The oncolytic herpes simplex virus vector, G47Δ, effectively targets tamoxifen-resistant breast cancer cells. Oncol. Rep. 2015, 35, 1741–1749. [Google Scholar] [CrossRef]
- Sahin, T.T.; Kasuya, H.; Nomura, N.; Shikano, T.; Yamamura, K.; Gewen, T.; Kanzaki, A.; Fujii, T.; Sugae, T.; Imai, T.; et al. Impact of novel oncolytic virus HF10 on cellular components of the tumor microenviroment in patients with recurrent breast cancer. Cancer Gene Ther. 2011, 19, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.; Winder, N.J.; Atkinson, E.; Al-Janabi, H.H.; Allen, R.J.; Hughes, R.; Moamin, M.R.; Louie, R.; Evans, D.; Hutchinson, M.; et al. Macrophages mediate the anti-tumor effects of the oncolytic virus HSV1716 in mammary tumors. Mol. Cancer Ther. 2020, 20, 589–601. [Google Scholar] [CrossRef]
- Marcato, P.; A Dean, C.; A Giacomantonio, C.; Lee, P.W. Oncolytic reovirus effectively targets breast cancer stem cells. Mol. Ther. 2009, 17, 972–979. [Google Scholar] [CrossRef]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef] [Green Version]
- Comins, C.; Spicer, J.; Protheroe, A.; Roulstone, V.; Twigger, K.; White, C.M.; Vile, R.; Melcher, A.; Coffey, M.C.; Mettinger, K.L.; et al. REO-10: A phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin. Cancer Res. 2010, 16, 5564–5572. [Google Scholar] [CrossRef] [Green Version]
- Lolkema, M.P.; Arkenau, H.T.; Harrington, K.; Roxburgh, P.; Morrison, R.; Roulstone, V.; Twigger, K.; Coffey, M.; Mettinger, K.; Gill, G.; et al. A phase I study of the com-bination of intravenous reovirus type 3 Dearing and gemcitabine in patients with advanced cancer. Clin. Cancer Res. 2011, 17, 581–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafa, A.A.; Meyers, D.E.; Thirukkumaran, C.M.; Liu, P.J.; Gratton, K.; Spurrell, J.; Shi, Q.; Thakur, S.; Morris, D.G. Oncolytic reovirus and immune checkpoint inhibition as a novel immunotherapeutic strategy for breast cancer. Cancers 2018, 10, 205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene laherparepvec improves durable response rate in patients with advanced melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Cripe, T.P.; Ngo, M.C.; Geller, J.I.; Louis, C.U.; Currier, M.A.; Racadio, J.M.; Towbin, A.; Rooney, C.M.; Pelusio, A.; Moon, A.; et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol. Ther. 2015, 23, 602–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Karapanagiotou, E.M.; Roulstone, V.; Twigger, K.; Ball, M.; Tanay, M.; Nutting, C.; Newbold, K.; Gore, M.E.; Larkin, J.; Syrigos, K.N.; et al. Phase I/II Trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin. Cancer Res. 2012, 18, 2080–2089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, V.; Ellard, S.L.; Dent, S.F.; Tu, D.; Mates, M.; Dhesy-Thind, S.K.; Panasci, L.; Gelmon, K.A.; Salim, M.; Song, X.; et al. A randomized phase II study of weekly paclitaxel with or without pelareorep in patients with metastatic breast cancer: Final analysis of Canadian Cancer Trials Group IND.213. Breast Cancer Res. Treat. 2018, 167, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.T.; Muñoz, L.E.; Stewart, R.M.R.; Selvaraj, P.; Mainou, B.A. Doxorubicin conjugation to reovirus improves oncolytic efficacy in triple-negative breast cancer. Mol. Ther. Oncolytics 2020, 18, 556–572. [Google Scholar] [CrossRef]
- O’Cathail, S.M.; Pokrovska, T.D.; Maughan, T.S.; Fisher, K.; Seymour, L.W.; Hawkins, M.A. Combining oncolytic adenovirus with radiation—A paradigm for the future of radiosensitization. Front. Oncol. 2017, 7, 153. [Google Scholar] [CrossRef]
- Wang, S.; Jhawar, S.; Bommareddy, P.; Thandoni, A.; Aspromonte, S.; Pepe, R.; Schiff, D.; Kaufman, H.; Goyal, S.; Goydos, J.; et al. Combined radiation and oncolytic viral therapy augments cytotoxic and immunogenic antitumor effects against melanoma. Int. J. Radiat. Oncol. 2018, 102, S153–S154. [Google Scholar] [CrossRef]
- O’Cathail, S.M.; Davis, S.; Holmes, J.; Brown, R.; Fisher, K.; Seymour, L.; Adams, R.; Good, J.; Sebag-Montefiore, D.; Maughan, T.; et al. A phase 1 trial of the safety, tolerability and biological effects of intravenous Enadenotucirev, a novel oncolytic virus, in combination with chemoradiotherapy in locally advanced rectal cancer (CEDAR). Radiat. Oncol. 2020, 15, 1–8. [Google Scholar] [CrossRef]
- Bieler, A.; Mantwill, K.; Holzmüller, R.; Jürchott, K.; Kaszubiak, A.; Stark, S.; Glockzin, G.; Lage, H.; Grosu, A.-L.; Gänsbacher, B.; et al. Impact of radiation therapy on the oncolytic adenovirus dl520: Implications on the treatment of glioblastoma. Radiother. Oncol. 2008, 86, 419–427. [Google Scholar] [CrossRef]
- Bourgeois-Daigneault, M.-C.; Roy, D.G.; Aitken, A.S.; El Sayes, N.; Martin, N.T.; Varette, O.; Falls, T.; St-Germain, L.; Pelin, A.; Lichty, B.D.; et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci. Transl. Med. 2018, 10, eaao1641. [Google Scholar] [CrossRef] [Green Version]
- Chaurasiya, S.; Yang, A.; Kang, S.; Lu, J.; Kim, S.-I.; Park, A.K.; Sivanandam, V.; Zhang, Z.; Woo, Y.; Warner, S.G.; et al. Oncolytic poxvirus CF33-hNIS-ΔF14.5 favorably modulates tumor immune microenvironment and works synergistically with anti-PD-L1 antibody in a triple-negative breast cancer model. OncoImmunology 2020, 9, 1729300. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.-Y. An engineered oncolytic virus expressing PD-L1 inhibitors activates tumor neoantigen-specific T cell responses. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Frazier, V.N.; Holl, E.; Brown, M.; Boczkowski, D.; Landa, K.; Hwang, S.; Gromeier, M.; Nair, S.K. Oncolytic poliovirus immunotherapy for breast cancer. J. Immunol. 2020, 204, 249.25. [Google Scholar]
- Matsumura, Y.; Maeda, H. A new concept for macromolecular therapeutics in cancer chemotherapy: Mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Danhier, F. To exploit the tumor microenvironment: Since the EPR effect fails in the clinic, what is the future of nanomedicine? J. Control. Release 2016, 244, 108–121. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Nace, R.; Ayala-Breton, C.; Steele, M.; Bailey, K.; Peng, K.W.; Russell, S.J. Perfusion pressure is a critical determinant of the intratumoral extravasation of oncolytic viruses. Mol. Ther. 2016, 24, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shee, K.; Yang, W.; Hinds, J.W.; Hampsch, R.A.; Varn, F.S.; Traphagen, N.A.; Patel, K.; Cheng, C.; Jenkins, N.P.; Kettenbach, A.N.; et al. Therapeutically targeting tumor micro-environment-mediated drug resistance in estrogen receptor-positive breast cancer. J. Exp. Med. 2018, 215, 895–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morrison, J.; Briggs, S.S.; Green, N.; Fisher, K.; Subr, V.; Ulbrich, K.; Kehoe, S.; Seymour, L.W. Virotherapy of ovarian cancer with polymer-cloaked adenovirus retargeted to the epidermal growth factor receptor. Mol. Ther. 2008, 16, 244–251. [Google Scholar] [CrossRef]
- Nosaki, K.; Hamada, K.; Takashima, Y.; Sagara, M.; Matsumura, Y.; Miyamoto, S.; Hijikata, Y.; Okazaki, T.; Nakanishi, Y.; Tani, K. A novel, polymer-coated oncolytic measles virus overcomes immune suppression and induces robust antitumor activity. Mol. Ther. Oncolytics 2016, 3, 16022. [Google Scholar] [CrossRef] [PubMed]
- Howard, F.; Muthana, M. Designer nanocarriers for navigating the systemic delivery of oncolytic viruses. Nanomedicine 2020, 15, 93–110. [Google Scholar] [CrossRef]
- Iscaro, A.; Howard, N.F.; Muthana, M. Nanoparticles: Properties and applications in cancer immunotherapy. Curr. Pharm. Des. 2019, 25, 1962–1979. [Google Scholar] [CrossRef]
- Muthana, M.; Giannoudis, A.; Scott, S.; Fang, H.-Y.; Coffelt, S.B.; Morrow, F.J.; Murdoch, C.; Burton, J.; Cross, N.; Burke, B.; et al. Use of macrophages to target therapeutic adenovirus to human prostate tumors. Cancer Res. 2011, 71, 1805–1815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamano, T.; Kubo, S.; Fukomoto, M.; Yano, A.; Mawatari-Furukawa, Y.; Okamura, H.; Tomita, N. Whole cell vaccination using immunogenic cell death by an oncolytic adenovirus is effective against a colorectal cancer model. Mol. Ther. Oncolytics 2016, 3, 16031. [Google Scholar] [CrossRef] [PubMed]
- Melzer, M.K.; Zeitlinger, L.; Mall, S.; Steiger, K.; Schmid, R.M.; Ebert, O.; Krackhardt, A.; Altomonte, J. Enhanced safety and efficacy of oncolytic VSV therapy by combination with T cell receptor transgenic T cells as carriers. Mol. Ther. Oncolytics 2019, 12, 26–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenna, M.K.; Englisch, A.; Brenner, B.; Smith, T.; Hoyos, V.; Suzuki, M.; Brenner, M.K. Mesenchymal stromal cell delivery of oncolytic immunotherapy improves CAR-T cell antitumor activity. Mol. Ther. 2021, 29, 1808–1820. [Google Scholar] [CrossRef]
- Lara, H.H.; Ayala-Nuñez, N.V.; Ixtepan-Turrent, L.; Rodriguez-Padilla, C. Mode of antiviral action of silver nanoparticles against HIV-1. J. Nanobiotechnology 2010, 8, 1. [Google Scholar] [CrossRef] [PubMed]
- Almstätter, I.; Mykhaylyk, O.; Settles, M.; Altomonte, J.; Aichler, M.; Walch, A.; Rummeny, E.J.; Ebert, O.; Plank, C.; Braren, R. Characterization of magnetic viral complexes for targeted delivery in oncology. Theranostics 2015, 5, 667–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tresilwised, N.; Pithayanukul, P.; Mykhaylyk, O.; Holm, P.S.; Holzmüller, R.; Anton, M.; Thalhammer, S.; Adigüzel, D.; Döblinger, M.; Plank, C. Boosting oncolytic adenovirus potency with magnetic nanoparticles and magnetic force. Mol. Pharm. 2010, 7, 1069–1089. [Google Scholar] [CrossRef]
- Muthana, M.; Kennerley, A.J.; Hughes, R.; Fagnano, E.; Richardson, J.; Paul, M.; Murdoch, C.; Wright, F.; Payne, C.; Lythgoe, M.; et al. Directing cell therapy to anatomic target sites in vivo with magnetic resonance targeting. Nat. Commun. 2015, 6, 8009. [Google Scholar] [CrossRef] [Green Version]
- Smida, T.; Bruno, T.C.; Stabile, L.P. Influence of estrogen on the NSCLC microenvironment: A comprehensive picture and clinical implications. Front. Oncol. 2020, 10, 137. [Google Scholar] [CrossRef] [Green Version]
- Ye, Y.; Jing, Y.; Li, L.; Mills, G.B.; Diao, L.; Liu, H.; Han, L. Sex-associated molecular differences for cancer immunotherapy. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svoronos, N.; Perales-Puchalt, A.; Allegrezza, M.J.; Rutkowski, M.R.; Payne, K.K.; Tesone, A.J.; Nguyen, J.M.; Curiel, T.J.; Cadungog, M.G.; Singhal, S.; et al. Tumor cell-independent estrogen signaling drives disease progression through mobilization of myeloid-derived suppressor cells. Cancer Discov. 2017, 7, 72–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Lara, V.; Hernandez-Martinez, J.-M.; Arrieta, O. Influence of estrogen in non-small cell lung cancer and its clinical implications. J. Thorac. Dis. 2018, 10, 482–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Márquez-Garbán, D.C.; Deng, G.; Comin-Anduix, B.; Garcia, A.J.; Xing, Y.; Chen, H.-W.; Cheung-Lau, G.; Hamilton, N.; Jung, M.E.; Pietras, R.J. Antiestrogens in combination with immune checkpoint inhibitors in breast cancer immunotherapy. J. Steroid Biochem. Mol. Biol. 2019, 193, 105415. [Google Scholar] [CrossRef]
- Özdemir, B.C.; Dotto, G.-P. Sex hormones and anticancer immunity. Clin. Cancer Res. 2019, 25, 4603–4610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stiles, B.M.; Adusumilli, P.S.; Stanziale, S.F.; Eisenberg, D.P.; Bhargava, A.; Kim, T.H.; Chan, M.-K.; Huq, R.; Gonen, M.; Fong, Y. Estrogen enhances the efficacy of an oncolytic HSV-1 mutant in the treatment of estrogen receptor-positive breast cancer. Int. J. Oncol. 2006, 28, 1429–1439. [Google Scholar] [CrossRef] [Green Version]
- Klein, S.L.; Flanagan, K.L. Sex differences in immune responses. Nat. Rev. Immunol. 2016, 16, 626–638. [Google Scholar] [CrossRef]
- Montecino-Rodriguez, E.; Berent-Maoz, B.; Dorshkind, K. Causes, consequences, and reversal of immune system aging. J. Clin. Investig. 2013, 123, 958–965. [Google Scholar] [CrossRef] [PubMed]
- Leng, J.; Goldstein, D.R. Impact of aging on viral infections. Microbes Infect. 2010, 12, 1120–1124. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Huang, M.; Kon, J.; Patel, K.; Schwartz, J.B.; Fast, K.; Ferrucci, L.; Madara, K.; Taub, D.D.; Longo, D.L. Gender specificity of altered human immune cytokine profiles in aging. FASEB J. 2010, 24, 3580–3589. [Google Scholar] [CrossRef] [Green Version]
- Kumru, S.; Godekmerdan, A.; Yilmaz, B. Immune effects of surgical menopause and estrogen replacement therapy in peri-menopausal women. J. Reprod. Immunol. 2004, 63, 31–38. [Google Scholar] [CrossRef]
- Giglio, T.; Imro, M.; Filaci, G.; Scudeletti, M.; Puppo, F.; De Cecco, L.; Indiveri, F.; Costantini, S. Immune cell circulating subsets are affected by gonadal function. Life Sci. 1994, 54, 1305–1312. [Google Scholar] [CrossRef]
- Cronin, K.A.; Harlan, L.C.; Dodd, K.W.; Abrams, J.S.; Ballard-Barbash, R. Population-based Estimate of the Prevalence of HER-2 Positive Breast Cancer Tumors for Early Stage Patients in the US. Cancer Investig. 2010, 28, 963–968. [Google Scholar] [CrossRef] [Green Version]
- Morse, M.A.; Hobeika, A.; Osada, T.; Niedzwiecki, D.; Marcom, P.K.; Blackwell, K.L.; Anders, C.; Devi, G.R.; Lyerly, H.K.; Clay, T.M. Long term disease-free survival and T cell and antibody responses in women with high-risk Her2+ breast cancer following vaccination against Her2. J. Transl. Med. 2007, 5, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liikanen, I.; Tähtinen, S.; Guse, K.; Gutmann, T.; Savola, P.; Oksanen, M.; Kanerva, A.; Hemminki, A. Oncolytic adenovirus expressing monoclonal antibody trastuzumab for treatment of HER2-positive cancer. Mol. Cancer Ther. 2016, 15, 2259–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| OV | Checkpoint Inhibitor | Cancer | Reference |
|---|---|---|---|
| Oncolytic vaccinia virus co-expressing a mouse PD-L1 inhibitor and GM-CSF. | PD-L1 inhibitor | Py230 breast cancer | [43] |
| Oncolytic reovirus—non-modified | PD-1 inhibitor | Immunocompetent, syngeneic EMT6 | [30] |
| Polio:rhinovirus recombinant (PVSRIPO) | PD1/PD-L1 axis | E0771 | [44] |
| Marabavirus—non-modified | Anti-PD-1 (clone RMPI-14, BioXCell) and anti-CTLA4 (clone 9D9, BioXCell) | EMT6, E0771, 4T1 immunocompetent, syngeneic neoadjuvant models | [41] |
| Modified measles virus (MV-NAP) | PD-1/PD-L1 blockade | Phase 1 trial ongoing (Mayo Clinic Breast Cancer SPORE) | https://grantome.com/grant/NIH/P50-CA116201-12 (accessed on: 1 May 2021) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kwan, A.; Winder, N.; Muthana, M. Oncolytic Virotherapy Treatment of Breast Cancer: Barriers and Recent Advances. Viruses 2021, 13, 1128. https://doi.org/10.3390/v13061128
Kwan A, Winder N, Muthana M. Oncolytic Virotherapy Treatment of Breast Cancer: Barriers and Recent Advances. Viruses. 2021; 13(6):1128. https://doi.org/10.3390/v13061128
Chicago/Turabian StyleKwan, Amy, Natalie Winder, and Munitta Muthana. 2021. "Oncolytic Virotherapy Treatment of Breast Cancer: Barriers and Recent Advances" Viruses 13, no. 6: 1128. https://doi.org/10.3390/v13061128
APA StyleKwan, A., Winder, N., & Muthana, M. (2021). Oncolytic Virotherapy Treatment of Breast Cancer: Barriers and Recent Advances. Viruses, 13(6), 1128. https://doi.org/10.3390/v13061128