
Spatial and Temporal Dynamics and Molecular Evolution of Tula orthohantavirus in German Vole Populations

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Rodent Trapping and Sample Collection
2.2. Nucleic Acid Isolation
2.3. Molecular Species and Sex Determination
2.4. TULV Detection
2.5. Sequence Determination and Phylogenetic Analyses
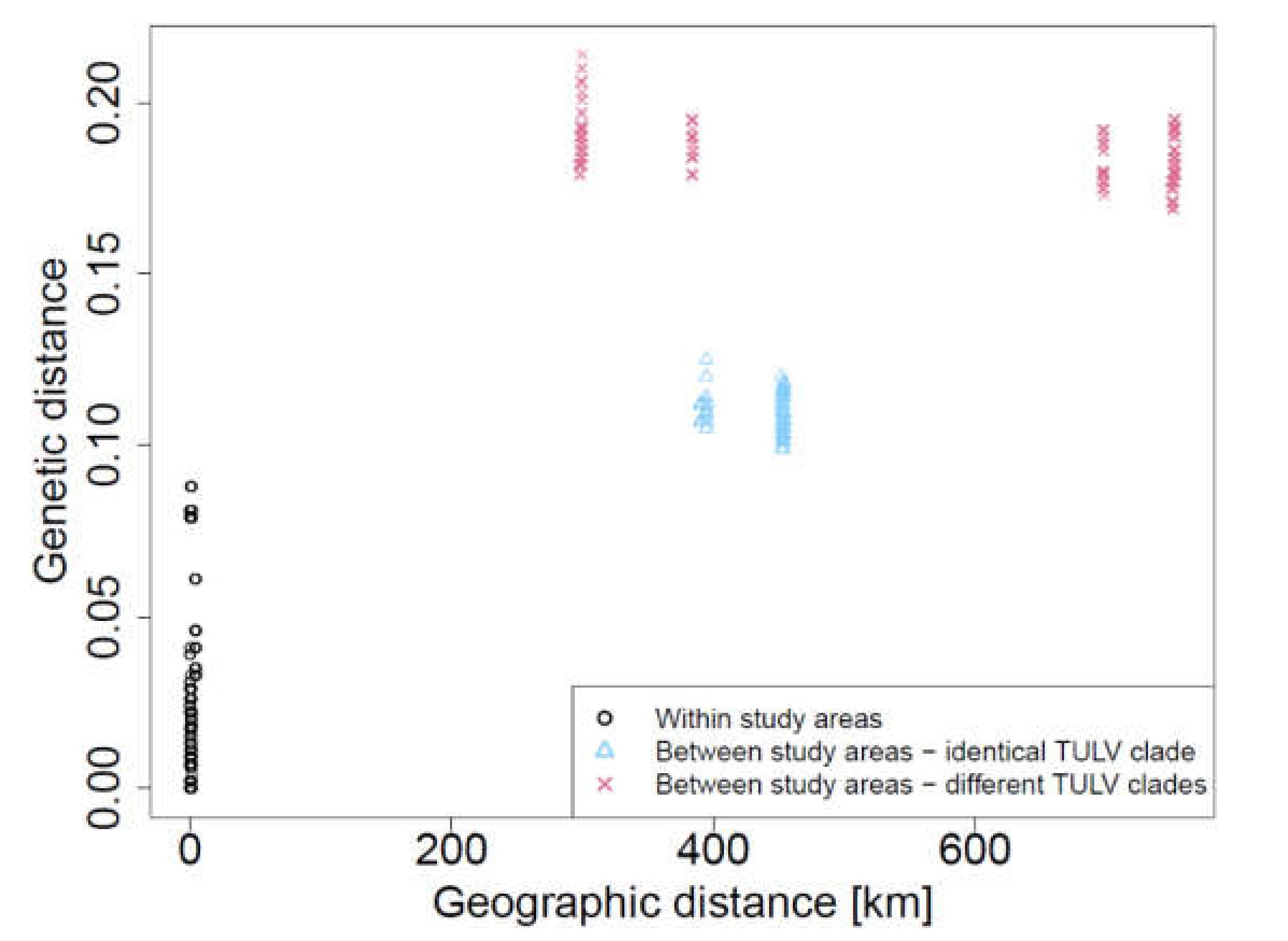
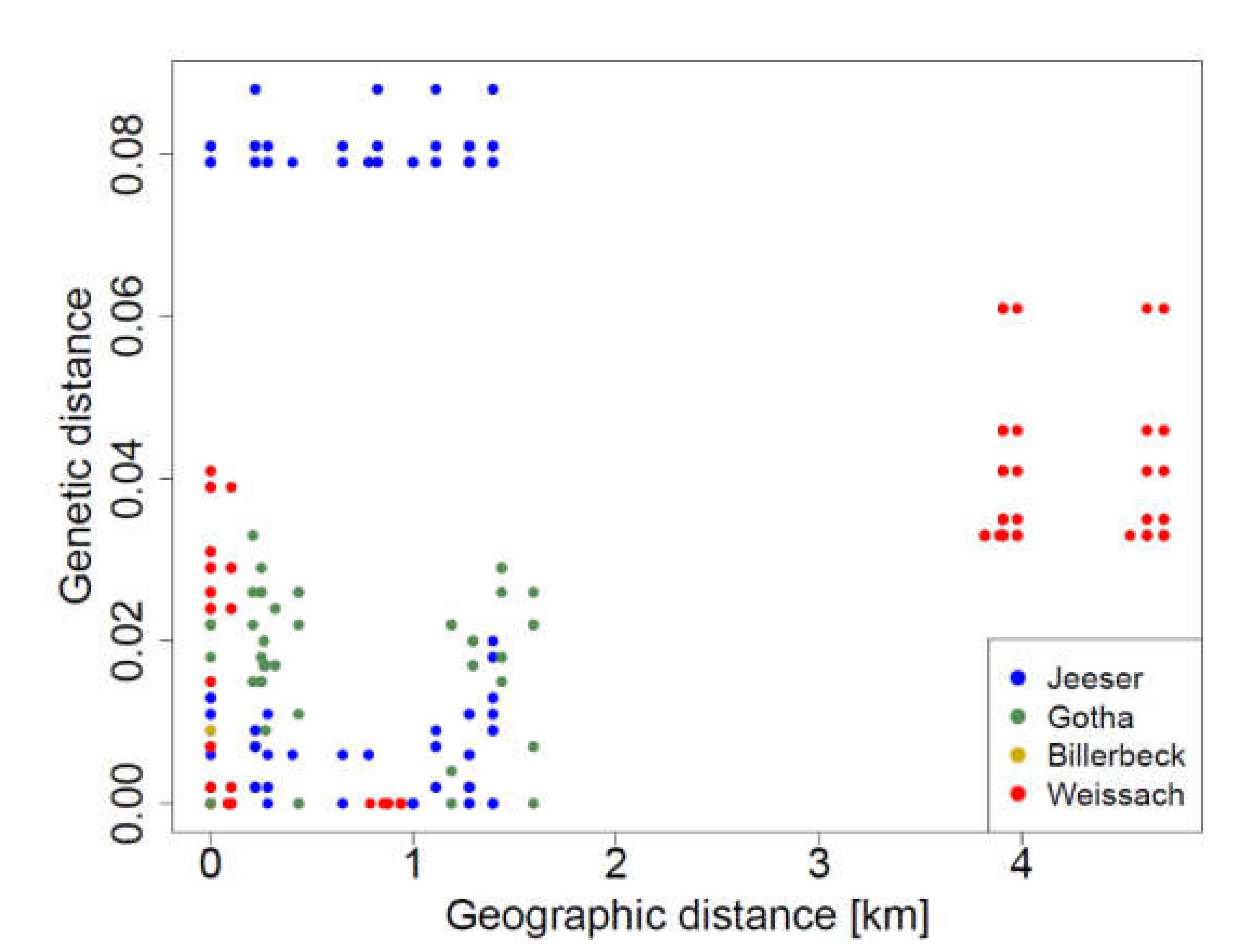
2.6. Isolation-by-Distance Analysis
2.7. Statistical Analysis
3. Results
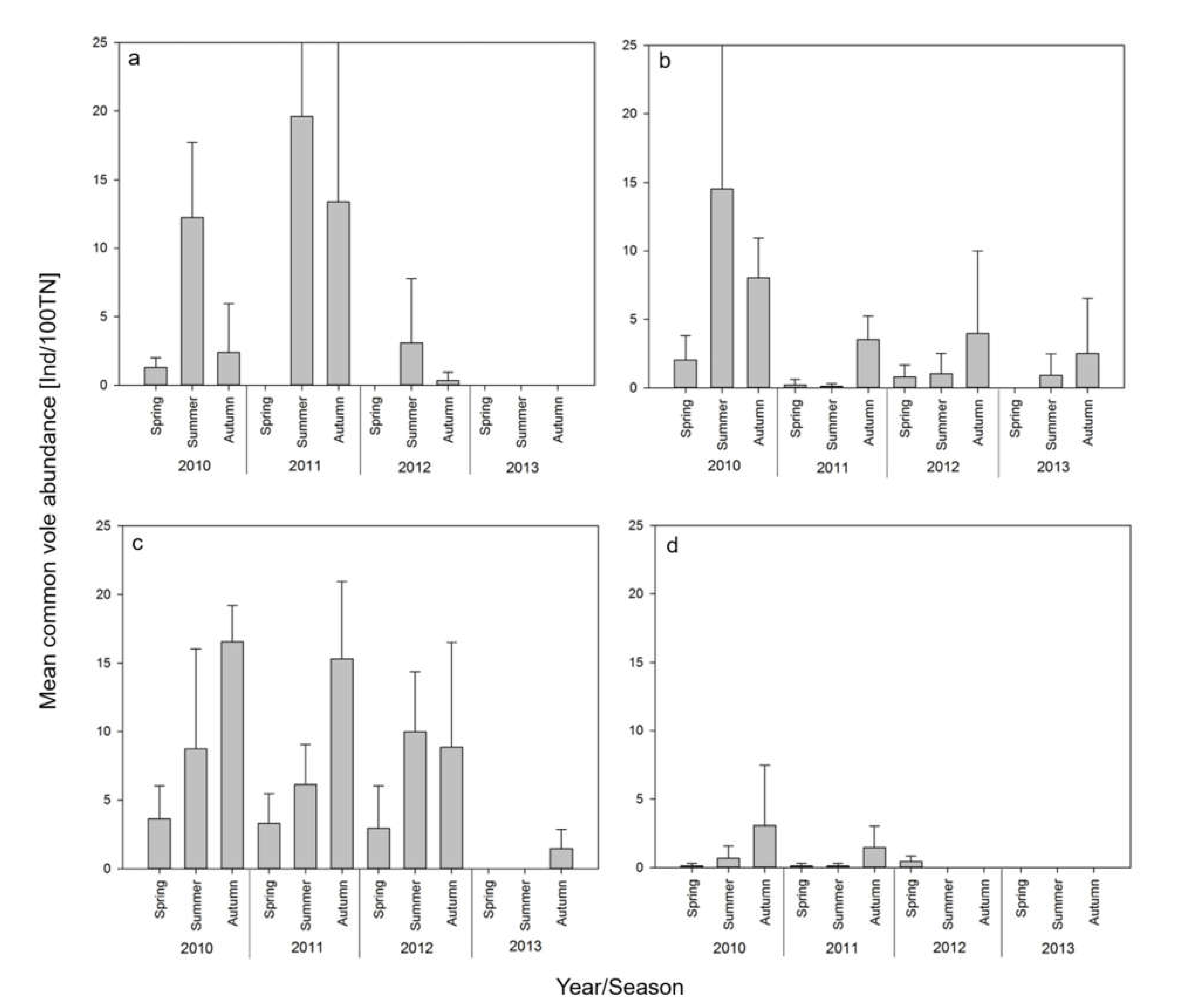
3.1. Rodent Trapping
3.2. TULV Seroprevalence
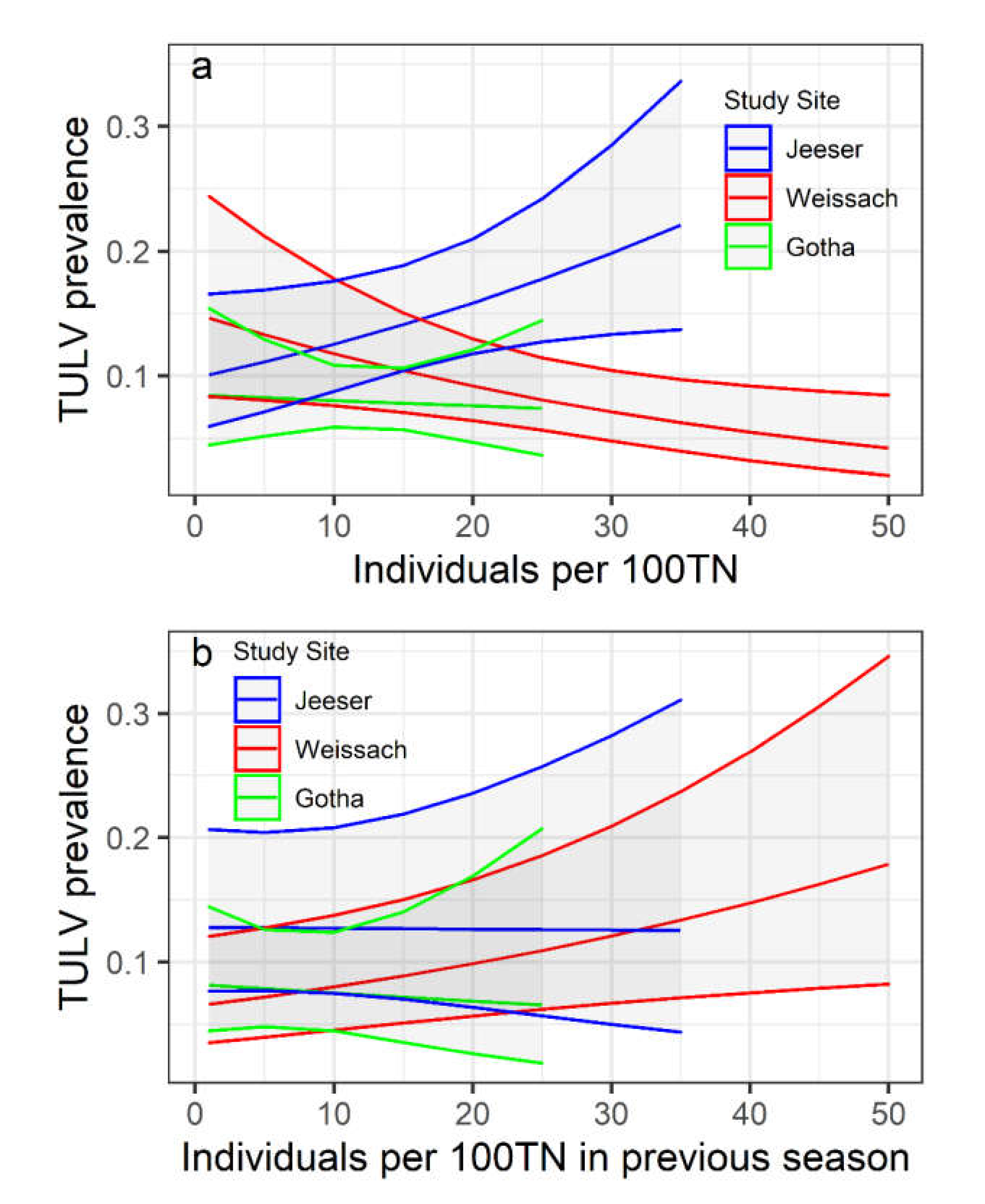
3.3. Relationship of TULV Seroprevalence with Common Vole Abundance
3.4. Detection of TULV RNA and Sequence Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Plyusnin, A.; Vapalahti, O.; Lankinen, H.; Lehväslaiho, H.; Apekina, N.; Myasnikov, Y.; Kallio-Kokko, H.; Henttonen, H.; Lundkvist, A.; Brummer-Korvenkontio, M.; et al. Tula virus: A newly detected hantavirus carried by European common voles. J. Virol. 1994, 68, 7833–7839. [Google Scholar] [CrossRef] [Green Version]
- Sibold, C.; Sparr, S.; Schulz, A.; Labuda, M.; Kozuch, O.; Lysỳ, J.; Krüger, D.H.; Meisel, H. Genetic characterization of a new hantavirus detected in Microtus arvalis from Slovakia. Virus Genes 1995, 10, 277–281. [Google Scholar] [CrossRef]
- Iakimenko, V.V.; Dekonenko, A.E.; Mal’kova, M.G.; Kuz’min, I.V.; Tantsev, A.K.; Dzagurova, T.K.; Tkachenko, E.A. The spread of hantaviruses in Western Siberia. Med. Parazitol. 2000, 3, 21–28. [Google Scholar]
- Scharninghausen, J.J.; Pfeffer, M.; Meyer, H.; Davis, D.S.; Honeycutt, R.L.; Faulde, M. Genetic evidence for Tula virus in Microtus arvalis and Microtus agrestis populations in Croatia. Vector Borne Zoonotic Dis. 2002, 2, 19–27. [Google Scholar] [CrossRef]
- Song, J.-W.; Gligic, A.; Yanagihara, R. Identification of Tula hantavirus in Pitymys subterraneus captured in the Cacak region of Serbia-Yugoslavia. Int. J. Infect. Dis. 2002, 6, 31–36. [Google Scholar] [CrossRef] [Green Version]
- Schmidt-Chanasit, J.; Essbauer, S.; Petraityte, R.; Yoshimatsu, K.; Tackmann, K.; Conraths, F.J.; Sasnauskas, K.; Arikawa, J.; Thomas, A.; Pfeffer, M.; et al. Extensive host sharing of Central European Tula virus. J. Virol. 2010, 84, 459–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlegel, M.; Kindler, E.; Essbauer, S.S.; Wolf, R.; Thiel, J.; Groschup, M.H.; Heckel, G.; Oehme, R.M.; Ulrich, R.G. Tula virus infections in the Eurasian water vole in Central Europe. Vector Borne Zoonotic Dis. 2012, 12, 503–513. [Google Scholar] [CrossRef] [Green Version]
- Tkachenko, E.A.; Witkowski, P.T.; Radosa, L.; Dzagurova, T.K.; Okulova, N.M.; Yunicheva, Y.V.; Vasilenko, L.; Morozov, V.G.; Malkin, G.A.; Krüger, D.H.; et al. Adler hantavirus, a new genetic variant of Tula virus identified in major’s pine voles (Microtus majori) sampled in Southern European Russia. Infect. Genet. Evol. 2015, 29, 156–163. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Saxenhofer, M.; Drewes, S.; Schlegel, M.; Wanka, K.M.; Frank, R.; Klimpel, S.; Von Blanckenhagen, F.; Maaz, D.; Herden, C.; et al. High genetic structuring of Tula hantavirus. Arch. Virol. 2016, 161, 1135–1149. [Google Scholar] [CrossRef]
- Hörling, J.; Chizhikov, V.; Lundkvist, A.; Jonsson, M.; Ivanov, L.; Dekonenko, A.; Niklasson, B.; Dzagurova, T.; Peters, C.J.; Tkachenko, E.; et al. Khabarovsk virus: A phylogenetically and serologically distinct hantavirus isolated from Microtus fortis trapped in Far-East Russia. J. Gen. Virol. 1996, 77, 687–694. [Google Scholar] [CrossRef] [PubMed]
- Kariwa, H.; Yoshimatsu, K.; Sawabe, J.; Yokota, E.; Arikawa, J.; Takashima, I.; Fukushima, H.; Lundkvist, Å.; Shubin, F.; Isachkova, L.M.; et al. Genetic diversities of hantaviruses among rodents in Hokkaido, Japan and Far East Russia. Virus Res. 1999, 59, 219–228. [Google Scholar] [CrossRef]
- Pounder, K.C.; Begon, M.; Sironen, T.; Henttonen, H.; Watts, P.C.; Voutilainen, L.; Vapalahti, O.; Klempa, B.; Fooks, A.R.; McElhinney, L.M. Novel hantavirus in wildlife, United Kingdom. Emerg. Infect. Dis. 2013, 19, 673–675. [Google Scholar] [CrossRef] [PubMed]
- Jeske, K.; Hiltbrunner, M.; Drewes, S.; Ryll, R.; Wenk, M.; Špakova, A.; Petraitytė-Burneikienė, R.; Heckel, G.; Ulrich, R.G. Field vole-associated Traemmersee hantavirus from Germany represents a novel hantavirus species. Virus Genes 2019, 55, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Drewes, S.; Jeske, K.; Straková, P.; Balčiauskas, L.; Ryll, R.; Balčiauskienė, L.; Kohlhause, D.; Schnidrig, G.-A.; Hiltbrunner, M.; Špakova, A.; et al. Identification of a novel hantavirus strain in the root vole (Microtus oeconomus) in Lithuania, Eastern Europe. Infect. Genet. Evol. 2020, 90, 104520. [Google Scholar] [CrossRef] [PubMed]
- Jääskeläinen, K.M.; Kaukinen, P.; Minskaya, E.S.; Plyusnina, A.; Vapalahti, O.; Elliott, R.M.; Weber, F.; Vaheri, A.; Plyusnin, A. Tula and Puumala hantavirus NSs ORFs are functional and the products inhibit activation of the interferon-beta promoter. J. Med. Virol. 2007, 79, 1527–1536. [Google Scholar] [CrossRef]
- Vapalahti, O.; Lundkvist, Å.; Kukkonen, S.K.; Cheng, Y.; Gilljam, M.; Kanerva, M.; Manni, T.; Pejcoch, M.; Niemimaa, J.; Kaikusalo, A.; et al. Isolation and characterization of Tula virus, a distinct serotype in the genus Hantavirus, family Bunyaviridae. J. Gen. Virol. 1996, 77, 3063–3067. [Google Scholar] [CrossRef]
- Saxenhofer, M.; Schmidt, S.; Ulrich, R.G.; Heckel, G. Secondary contact between diverged host lineages entails ecological speciation in a European hantavirus. PLoS Biol. 2019, 17, e3000142. [Google Scholar] [CrossRef] [Green Version]
- Hiltbrunner, M.; Heckel, G. Assessing genome-wide diversity in European hantaviruses through sequence capture from natural host samples. Viruses 2020, 12, 749. [Google Scholar] [CrossRef]
- Klempa, B.; Meisel, H.; Räth, S.; Bartel, J.; Ulrich, R.; Krüger, D.H. Occurrence of renal and pulmonary syndrome in a region of Northeast Germany where Tula hantavirus circulates. J. Clin. Microbiol. 2003, 41, 4894–4897. [Google Scholar] [CrossRef] [Green Version]
- Mertens, M.; Hofmann, J.; Petraityte-Burneikiene, R.; Ziller, M.; Sasnauskas, K.; Friedrich, R.; Niederstrasser, O.; Krüger, D.H.; Groschup, M.H.; Petri, E.; et al. Seroprevalence study in forestry workers of a non-endemic region in Eastern Germany reveals infections by Tula and Dobrava-Belgrade hantaviruses. Med. Microbiol. Immunol. 2011, 200, 263–268. [Google Scholar] [CrossRef]
- Zelená, H.; Mrázek, J.; Kuhn, T. Tula hantavirus infection in immunocompromised host, Czech Republic. Emerg. Infect. Dis. 2013, 19, 1873–1875. [Google Scholar] [CrossRef] [PubMed]
- Reynes, J.M.; Carli, D.; Boukezia, N.; Debruyne, M.; Herti, S. Tula hantavirus infection in a hospitalised patient, France, June 2015. Eurosurveillance 2015, 20, 30095. [Google Scholar] [CrossRef] [Green Version]
- Klempa, B. Dobrava and Tula hantaviruses from Central Europe: Molecular Evolution and Pathogenic Relevance. Ph.D. Thesis, Humboldt Universität zu Berlin, Berlin, Germany, 2004. [Google Scholar]
- Hofmann, J.; Kramer, S.; Herrlinger, K.R.; Jeske, K.; Kuhns, M.; Weiss, S.; Ulrich, R.G.; Krüger, D.H. Tula virus as causative agent of hantavirus disease in immunocompetent person, Germany. Emerg. Infect. Dis. 2021, 27, 1234–1237. [Google Scholar] [CrossRef] [PubMed]
- Mitchell-Jones, A.J.; Amori, G.; Bogdanowicz, W.; Krystufek, B.; Reijnders, P.J.H.; Spitzenberger, F.; Stubbe, M.; Thissen, J.B.M.; Vohralik, V.; Zima, J. The Atlas of European Mammals; Academic Press: London, UK, 1999. [Google Scholar]
- Jacob, J.; Manson, P.; Barfknecht, R.; Fredricks, T. Common vole (Microtus arvalis) ecology and management: Implications for risk assessment of plant protection products. Pest Manag. Sci. 2014, 70, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Imholt, C.; Esther, A.; Perner, J.; Jacob, J. Identification of weather parameters related to regional population outbreak risk of common voles (Microtus arvalis) in Eastern Germany. Wildl. Res. 2011, 38, 551–559. [Google Scholar] [CrossRef]
- Esther, A.; Imholt, C.; Perner, J.; Schumacher, J.; Jacob, J. Correlations between weather conditions and common vole (Microtus arvalis) densities identified by regression tree analysis. Basic Appl. Ecol. 2014, 15, 75–84. [Google Scholar] [CrossRef]
- Delattre, P.; De Sousa, B.; Fichet-Calvet, E.; Quere, J.P.; Giraudoux, P. Vole outbreaks in a landscape context: Evidence from a six year study of Microtus arvalis. Landsc. Ecol. 1999, 14, 401–412. [Google Scholar] [CrossRef]
- Bryja, J.; Tkadlec, E.; Nesvadbova, J.; Gaisler, J.; Zejda, J. Comparison of enumeration and Jolly-Seber estimation of population size in the common vole (Microtus arvalis). Acta Theriol. 2001, 46, 279–285. [Google Scholar] [CrossRef]
- Jacob, J.; Tkadlec, E. Rodent Outbreaks: Ecology and Impacts; International Rice Research Institute: Los Baños, Philippines, 2010. [Google Scholar]
- Jacob, J.; Imholt, C.; Caminero-Saldaña, C.; Couval, G.; Giraudoux, P.; Herrero-Cófreces, S.; Horváth, G.; Luque-Larena, J.J.; Tkadlec, E.; Wymenga, E. Europe-wide outbreaks of common voles in 2019. J. Pest Sci. 2020, 93, 703–709. [Google Scholar] [CrossRef] [Green Version]
- Kallio, E.R.; Begon, M.; Henttonen, H.; Koskela, E.; Mappes, T.; Vaheri, A.; Vapalahti, O. Cyclic hantavirus epidemics in humans—Predicted by rodent host dynamics. Epidemics 2009, 1, 101–107. [Google Scholar] [CrossRef]
- Kallio, E.R.; Begon, M.; Henttonen, H.; Koskela, E.; Mappes, T.; Vaheri, A.; Vapalahti, O. Hantavirus infections in fluctuating host populations: The role of maternal antibodies. Proc. R. Soc. B Biol. Sci. 2010, 277, 3783–3791. [Google Scholar] [CrossRef] [PubMed]
- Reil, D.; Rosenfeld, U.M.; Imholt, C.; Schmidt, S.; Ulrich, R.G.; Eccard, J.A.; Jacob, J. Puumala hantavirus infections in bank vole populations: Host and virus dynamics in Central Europe. BMC Ecol. 2017, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voutilainen, L.; Kallio, E.R.; Niemimaa, J.; Vapalahti, O.; Henttonen, H. Temporal dynamics of Puumala hantavirus infection in cyclic populations of bank voles. Sci. Rep. 2016, 6, 21323. [Google Scholar] [CrossRef] [Green Version]
- Weber de Melo, V.; Sheikh Ali, H.; Freise, J.; Kühnert, D.; Essbauer, S.; Mertens, M.; Wanka, K.M.; Drewes, S.; Ulrich, R.G.; Heckel, G. Spatiotemporal dynamics of Puumala hantavirus associated with its rodent host, Myodes glareolus. Evol. Appl. 2015, 8, 545–559. [Google Scholar] [CrossRef] [Green Version]
- Fischer, S.; Mayer-Scholl, A.; Imholt, C.; Spierling, N.G.; Heuser, E.; Schmidt, S.; Reil, D.; Rosenfeld, U.M.; Jacob, J.; Nöckler, K.; et al. Leptospira genomospecies and sequence type prevalence in small mammal populations in Germany. Vector Borne Zoonotic Dis. 2018, 18, 188–199. [Google Scholar] [CrossRef]
- Schlegel, M.; Sheikh Ali, H.; Keller, M.; Groschup, M.; Ulrich, R. Molekulare Artbestimmung von Kleinsäugern. LabLoeffler 2013, 7, 16–17. [Google Scholar]
- Niethammer, J.; Krapp, F. Handbuch der Säugetiere Europas; Akademische Verlagsgesellschaft: Leipzig, Germany, 2004; Volume 2. [Google Scholar]
- Schlegel, M.; Ali, H.S.; Stieger, N.; Groschup, M.H.; Wolf, R.; Ulrich, R.G. Molecular identification of small mammal species using novel cytochrome B gene-derived degenerated primers. Biochem. Genet. 2012, 50, 440–447. [Google Scholar] [CrossRef]
- Braaker, S.; Heckel, G. Transalpine colonisation and partial phylogeographic erosion by dispersal in the common vole (Microtus arvalis). Mol. Ecol. 2009, 18, 2518–2531. [Google Scholar] [CrossRef] [PubMed]
- Aasen, E.; Medrano, J.F. Amplification of the ZFY and ZFX genes for sex identification in humans, cattle, sheep and goats. Biotechnology 1990, 8, 1279–1281. [Google Scholar] [CrossRef]
- Bryja, J.; Konecny, A. Fast sex identification in wild mammals using PCR amplification of the Sry gene. FOLIA Zool. 2003, 52, 269–274. [Google Scholar]
- Essbauer, S.; Schmidt, J.; Conraths, F.J.; Friedrich, R.; Koch, J.; Hautmann, W.; Pfeffer, M.; Wölfel, R.; Finke, J.; Dobler, G.; et al. A new Puumala hantavirus subtype in rodents associated with an outbreak of Nephropathia epidemica in South-East Germany in 2004. Epidemiol. Infect. 2006, 134, 1333–1344. [Google Scholar] [CrossRef]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Denys, C.; Koivogui, L.; Ter Meulen, J.; Krüger, D.H. Hantavirus in African wood mouse, Guinea. Emerg. Infect. Dis. 2006, 12, 838–840. [Google Scholar] [CrossRef]
- GenBank, National Center for Biotechnology Information (NCBI). Available online: https://www.ncbi.nlm.nih.gov/genbank/ (accessed on 31 March 2021).
- Hall, T. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. The CIPRES science gateway: A community resource for phylogenetic analyses. In Extreme Digital Discovery, Proceedings of the 2011 TeraGrid Conference, Salt Lake City, UT, USA, 18–21 July 2011; Association for Computing Machinery: New York, NY, USA, 2011; pp. 1–8. [Google Scholar]
- Saxenhofer, M.; Weber de Melo, V.; Ulrich, R.G.; Heckel, G. Revised time scales of RNA virus evolution based on spatial information. Proc. R. Soc. B Biol. Sci. 2017, 284, 20170857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Hijmans, R.J. Geosphere: Spherical Trigonometry. R package version 1.5-10. 2019. Available online: https://CRAN.R-project.org/package=geosphere (accessed on 10 June 2021).
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020.
- Dray, S.; Dufour. A.B. The ade4 package: Implementing the duality diagram for ecologists. J. Stat. Softw. 2007, 22, 1–20. [Google Scholar] [CrossRef] [Green Version]
- Korner-Nievergelt, F.; Roth, T.; Von Felten, S.; Guelat, J.; Almasi, B.; Korner-Nievergelt, P. Bayesian Data Analysis in Ecology Using Linear Models with R, BUGS and Stan; Elsevier: Cambridge, MA, USA, 2015. [Google Scholar]
- Binder, F.; Ryll, R.; Drewes, S.; Jagdmann, S.; Reil, D.; Hiltbrunner, M.; Rosenfeld, U.M.; Imholt, C.; Jacob, J.; Heckel, G.; et al. Spatial and temporal evolutionary patterns in Puumala orthohantavirus (PUUV) S segment. Pathogens 2020, 9, 548. [Google Scholar] [CrossRef] [PubMed]
- Jeske, K.; Jacob, J.; Drewes, S.; Pfeffer, M.; Heckel, G.; Ulrich, R.G.; Imholt, C. Hantavirus-Leptospira coinfections in small mammals from Central Germany. Epidemiol. Infect. 2021, 149, e97. [Google Scholar] [CrossRef]
- Schweizer, M.; Excoffier, L.; Heckel, G. Fine-Scale genetic structure and dispersal in the common vole (Microtus arvalis). Mol. Ecol. 2007, 16, 2463–2473. [Google Scholar] [CrossRef]
- Schmidt, S.; Essbauer, S.S.; Mayer-Scholl, A.; Poppert, S.; Schmidt-Chanasit, J.; Klempa, B.; Henning, K.; Schares, G.; Groschup, M.H.; Spitzenberger, F.; et al. Multiple infections of rodents with zoonotic pathogens in Austria. Vector Borne Zoonotic Dis. 2014, 14, 467–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deter, J.; Chaval, Y.; Galan, M.; Gauffre, B.; Morand, S.; Henttonen, H.; Laakkonen, J.; Voutilainen, L.; Charbonnel, N.; Cosson, J.-F. Kinship, dispersal and hantavirus transmission in bank and common voles. Arch. Virol. 2008, 153, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Plyusnin, A.; Cheng, Y.; Vapalahti, O.; Pejcoch, M.; Unar, J.; Jelinkova, Z.; Lehväslaiho, H.; Lundkvist, Å.; Vaheri, A. Genetic variation in Tula hantaviruses: Sequence analysis of the S and M segments of strains from Central Europe. Virus Res. 1995, 39, 237–250. [Google Scholar] [CrossRef]
- Heroldová, M.; Pejčoch, M.; Bryja, J.; Jánová, E.; Suchomel, J.; Tkadlec, E. Tula virus in populations of small terrestrial mammals in a rural landscape. Vector Borne Zoonotic Dis. 2010, 10, 599–603. [Google Scholar] [CrossRef] [Green Version]
- Sibold, C.; Meisel, H.; Lundkvist, Å.; Schulz, A.; Cifire, F.; Ulrich, R.; Kozuch, O.; Labuda, M.; Krüger, D.H. Simultaneous occurrence of Dobrava, Puumala, and Tula hantaviruses in Slovakia. Am. J. Trop. Med. Hyg. 1999, 61, 409–411. [Google Scholar] [CrossRef] [Green Version]
- Heyman, P.; Klingström, J.; De Jaegere, F.; Leclercq, G.; Rozenfeld, F.; Escutenaire, S.; Vandenvelde, C.; Zizi, M.; Plyusnin, A.; Lundkvist, Å. Tula hantavirus in Belgium. Epidemiol. Infect. 2002, 128, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Plyusnina, A.; Laakkonen, J.; Niemimaa, J.; Henttonen, H.; Plyusnin, A. New genetic lineage of Tula hantavirus in Microtus Arvalis Obscurus in Eastern Kazakhstan. Open Virol. J. 2008, 2, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maas, M.; De Vries, A.; Van Roon, A.; Takumi, K.; Van der Giessen, J.; Rockx, B. High prevalence of Tula hantavirus in common voles in The Netherlands. Vector Borne Zoonotic Dis. 2017, 17, 200–205. [Google Scholar] [CrossRef]
- Hahne, J.; Jenkins, T.; Halle, S.; Heckel, G. Establishment success and resulting fitness consequences for vole dispersers. Oikos 2011, 120, 95–105. [Google Scholar] [CrossRef]
- Reijniers, J.; Tersago, K.; Borremans, B.; Hartemink, N.; Voutilainen, L.; Henttonen, H.; Leirs, H. Why hantavirus prevalence does not always increase with host density: Modeling the role of host spatial behavior and maternal antibodies. Front. Cell. Infect. Microbiol. 2020, 10, 536660. [Google Scholar] [CrossRef]
- Luis, A.D.; Douglass, R.J.; Mills, J.N.; Bjørnstad, O.N. Environmental fluctuations lead to predictability in Sin Nombre hantavirus outbreaks. Ecology 2015, 96, 1691–1701. [Google Scholar] [CrossRef]
- Hein, S.; Jacob, J. Population recovery of a common vole population (Microtus arvalis) after population collapse. Pest Manag. Sci. 2019, 75, 908–914. [Google Scholar] [CrossRef]
- Rodriguez-Pastor, R.; Jose Luque-Larena, J.; Lambin, X.; Mougeot, F. “Living on the edge”: The Role of field margins for common vole (Microtus arvalis) populations in recently colonised Mediterranean farmland. Agric. Ecosyst. Environ. 2016, 231, 206–217. [Google Scholar] [CrossRef] [Green Version]
- Carver, S.; Kuenzi, A.; Bagamian, K.H.; Mills, J.N.; Rollin, P.E.; Zanto, S.N.; Douglass, R. A temporal dilution effect: Hantavirus infection in deer mice and the intermittent presence of voles in Montana. Oecologia 2011, 166, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Muyangwa, M.; Martynova, E.V.; Khaiboullina, S.F.; Morzunov, S.P.; Rizvanov, A.A. Hantaviral proteins: Structure, functions, and role in hantavirus infection. Front. Microbiol. 2015, 6, 1326. [Google Scholar] [CrossRef] [PubMed]
- Handke, W.; Oelschlegel, R.; Franke, R.; Wiedemann, L.; Krüger, D.H.; Rang, A. Generation and characterization of genetic reassortants between Puumala and Prospect Hill hantavirus in vitro. J. Gen. Virol. 2010, 91, 2351–2359. [Google Scholar] [CrossRef] [PubMed]
- Kirsanovs, S.; Klempa, B.; Franke, R.; Lee, M.-H.; Schönrich, G.; Rang, A.; Kruger, D.H. Genetic reassortment between high-virulent and low-virulent Dobrava-Belgrade virus strains. Virus Genes 2010, 41, 319–328. [Google Scholar] [CrossRef]
- Klempa, B. Reassortment events in the evolution of hantaviruses. Virus Genes 2018, 54, 638–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weissach | Jeeser | Billerbeck | Gotha | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Year | Season | Site | Positive/ Total | % | Positive/ Total | % | Positive/ Total | % | Positive/ Total | % | Total % |
| 2010 | Spring | 1 | 1/6 | 16.7 | 0/9 | 0 | 0/0 | NA | 0/1 | NA | 2.8 |
| 2 | 0/0 | NA | 0/1 | NA | 0/0 | NA | 0/2 | NA | |||
| 3 | 0/5 | 0 | 0/3 | NA | 0/0 | NA | 0/9 | 0 | |||
| Summer | 1 | 6/47 | 12.8 | 1/12 | 8.3 | 0/0 | NA | 4/18 | 22 | 14.1 | |
| 2 | 3/18 | 16.7 | 6/24 | 25 | 0/1 | NA | 2/27 | 7.4 | |||
| 3 | 0/13 | 0 | 16/84 | 19 | 0/5 | 0 | 0/20 | 0 | |||
| Autumn | 1 | 0/0 | NA | 0/15 | 0 | 0/2 | NA | 2/35 | 5.7 | 6.8 | |
| 2 | 0/2 | NA | 3/22 | 14 | 2/18 | 11 | 1/41 | 2.4 | |||
| 3 | 4/17 | 23.5 | 5/30 | 17 | 0/0 | NA | 7/49 | 14 | |||
| 2011 | Spring | 1 | 0/0 | NA | 0/0 | NA | 0/0 | NA | 2/6 | 33 | 12.5 |
| 2 | 0/0 | NA | 0/0 | NA | 0/0 | NA | 1/16 | 6.3 | |||
| 3 | 0/0 | NA | 1/2 | NA | 0/1 | NA | 0/7 | 0 | |||
| Summer | 1 | 0/16 | 0 | 0/0 | NA | 0/0 | NA | 0/24 | 0 | 0.0 | |
| 2 | 0/17 | 0 | 0/0 | NA | 0/0 | NA | 0/11 | 0 | |||
| 3 | 0/103 | 0 | 0/0 | NA | 0/1 | NA | 0/14 | 0 | |||
| Autumn | 1 | 0/4 | NA | 1/10 | 10 | 0/0 | NA | 1/33 | 3 | 7.4 | |
| 2 | 0/0 | NA | 0/14 | 0 | 0/4 | NA | 3/60 | 5 | |||
| 3 | 12/110 | 10.9 | 0/4 | NA | 0/9 | 0 | 4/35 | 11 | |||
| 2012 | Spring | 1 | 0/0 | NA | 0/5 | 0 | 0/2 | NA | 2/16 | 13 | 11.4 |
| 2 | 0/0 | NA | 0/0 | NA | 0/2 | NA | 0/0 | NA | |||
| 3 | 0/0 | NA | 0/2 | NA | 0/0 | NA | 2/8 | 25 | |||
| Summer | 1 | 2/21 | 9.5 | 0/8 | 0 | 0/0 | NA | 2/29 | 6.9 | 9.1 | |
| 2 | 0/2 | NA | 0/0 | NA | 0/0 | NA | 2/14 | 14 | |||
| 3 | 0/0 | NA | 0/1 | NA | 0/0 | NA | 7/35 | 20 | |||
| Autumn | 1 | 0/0 | NA | 1/2 | NA | No Trapping | 0/30 | 0 | 12.0 | ||
| 2 | 0/0 | NA | 0/0 | NA | 0/1 | NA | |||||
| 3 | 0/3 | NA | 8/29 | 28 | 2/27 | 7.4 | |||||
| 2013 | Spring | 1 | No Trapping | 0/0 | NA | No Trapping | 0/0 | NA | 0.0 | ||
| 2 | 0/0 | NA | 0/0 | NA | |||||||
| 3 | 0/0 | NA | 0/0 | NA | |||||||
| Summer | 1 | No Trapping | 0/2 | NA | No Trapping | No Trapping | 0.0 | ||||
| 2 | 0/0 | NA | |||||||||
| 3 | 0/0 | NA | |||||||||
| Autumn | 1 | No Trapping | 0/0 | NA | No Trapping | 0/1 | NA | 3.6 | |||
| 2 | 0/1 | NA | 0/1 | NA | |||||||
| 3 | 1/21 | 4.8 | 0/4 | NA | |||||||
| Total | 28/384 | 7.3 | 42/301 | 14 | 2/45 | 4.4 | 44/574 | 7.7 | 10.4 | ||
| Same Season (Direct Effect) | Previous Season (Delayed Effect) | |||||||
|---|---|---|---|---|---|---|---|---|
| Parameter | Estimate | SE | z | p | Estimate | SE | z | p |
| Intercept | −1.735 | 0.333 | −5.215 | 0 | −2.675 | 0.344 | −7.785 | 0 |
| Abundance | −0.028 | 0.012 | −2.335 | 0.02 | 0.023 | 0.01 | 2.315 | 0.021 |
| Jeeser | −0.481 | 0.45 | −1.069 | 0.285 | 0.755 | 0.454 | 1.662 | 0.097 |
| Gotha | −0.643 | 0.5 | −1.286 | 0.198 | 0.261 | 0.49 | 0.531 | 0.595 |
| Abundance: Jeeser | 0.055 | 0.018 | 3.001 | 0.003 | −0.024 | 0.02 | −1.212 | 0.226 |
| Abundance: Gotha | 0.022 | 0.03 | 0.73 | 0.466 | −0.033 | 0.036 | −0.926 | 0.354 |
| Random factor | Variance | SD | Variance | SD | ||||
| Site:SA | 0 | 0 | 0.129 | 0.359 | ||||
| SA | 0 | 0 | 0 | 0 | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, S.; Reil, D.; Jeske, K.; Drewes, S.; Rosenfeld, U.M.; Fischer, S.; Spierling, N.G.; Labutin, A.; Heckel, G.; Jacob, J.; et al. Spatial and Temporal Dynamics and Molecular Evolution of Tula orthohantavirus in German Vole Populations. Viruses 2021, 13, 1132. https://doi.org/10.3390/v13061132
Schmidt S, Reil D, Jeske K, Drewes S, Rosenfeld UM, Fischer S, Spierling NG, Labutin A, Heckel G, Jacob J, et al. Spatial and Temporal Dynamics and Molecular Evolution of Tula orthohantavirus in German Vole Populations. Viruses. 2021; 13(6):1132. https://doi.org/10.3390/v13061132
Chicago/Turabian StyleSchmidt, Sabrina, Daniela Reil, Kathrin Jeske, Stephan Drewes, Ulrike M. Rosenfeld, Stefan Fischer, Nastasja G. Spierling, Anton Labutin, Gerald Heckel, Jens Jacob, and et al. 2021. "Spatial and Temporal Dynamics and Molecular Evolution of Tula orthohantavirus in German Vole Populations" Viruses 13, no. 6: 1132. https://doi.org/10.3390/v13061132
APA StyleSchmidt, S., Reil, D., Jeske, K., Drewes, S., Rosenfeld, U. M., Fischer, S., Spierling, N. G., Labutin, A., Heckel, G., Jacob, J., Ulrich, R. G., & Imholt, C. (2021). Spatial and Temporal Dynamics and Molecular Evolution of Tula orthohantavirus in German Vole Populations. Viruses, 13(6), 1132. https://doi.org/10.3390/v13061132