
TASPERT: Target-Specific Reverse Transcript Pools to Improve HTS Plant Virus Diagnostics

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
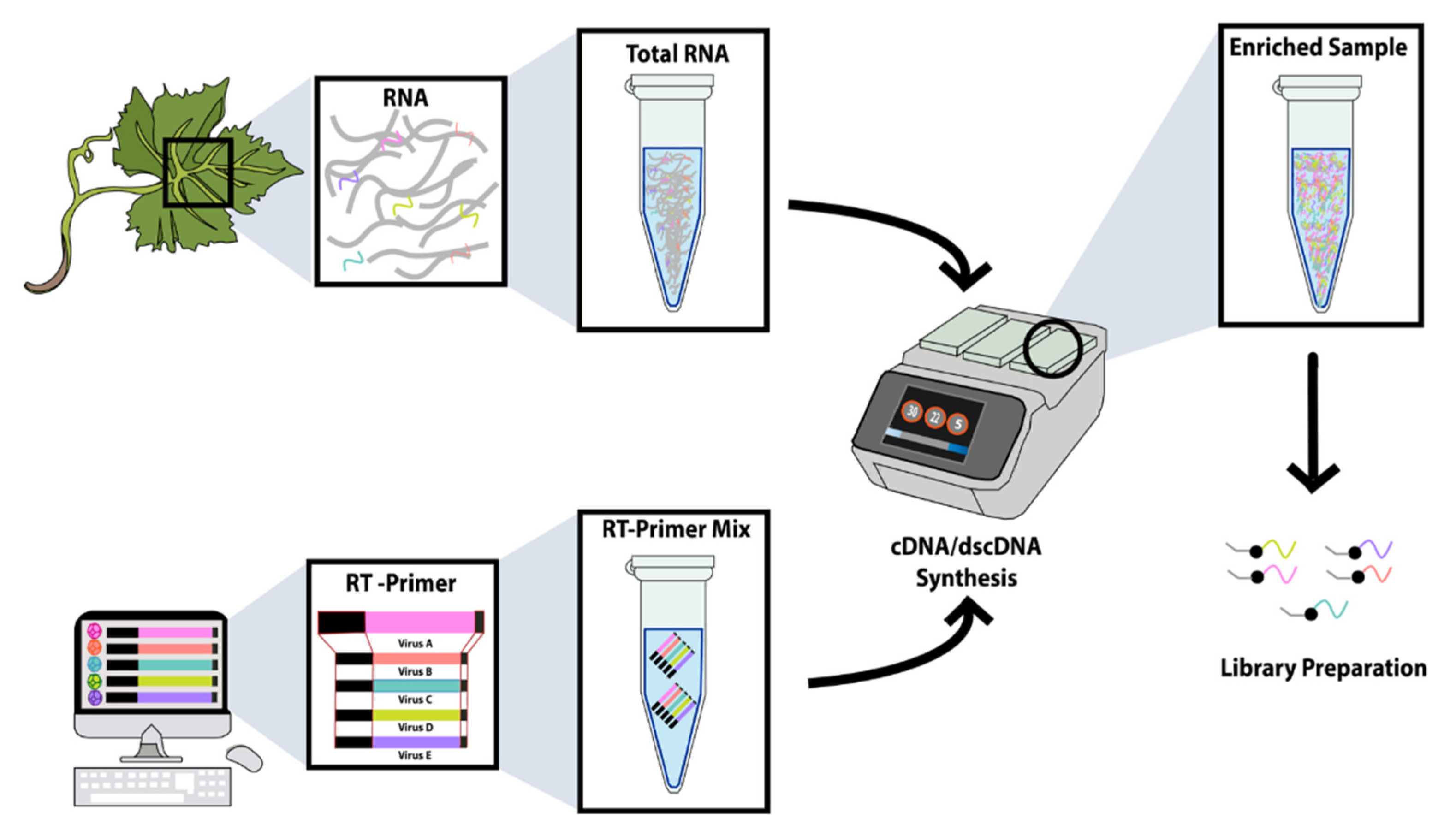
2.1. Experimental Design
2.2. Samples and RNA Extraction
2.3. Reverse Primer (RT Primer) Design at the 3′-Terminus for TASPERT
2.4. cDNA Synthesis and Amplification with TASPERT Pools
2.4.1. Assessing the Presence of TRSV and GLRaV3 in the ds-cDNA
2.4.2. Sequencing and HTS Diagnostics
3. Results
3.1. Reverse Primer (RT Primer) Design at the 3′-Terminus for TASPERT
3.2. Assessing the Presence of TRSV and GLRaV3 in the ds-cDNA
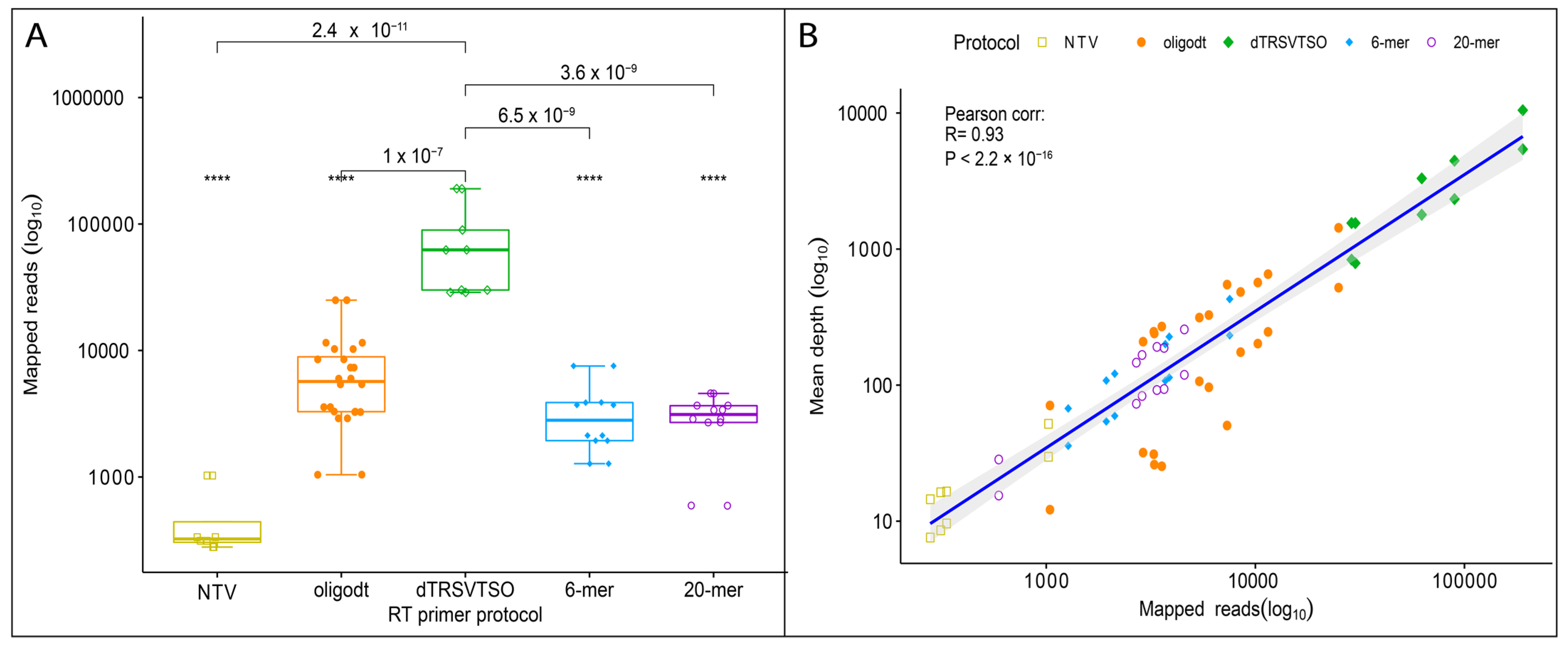
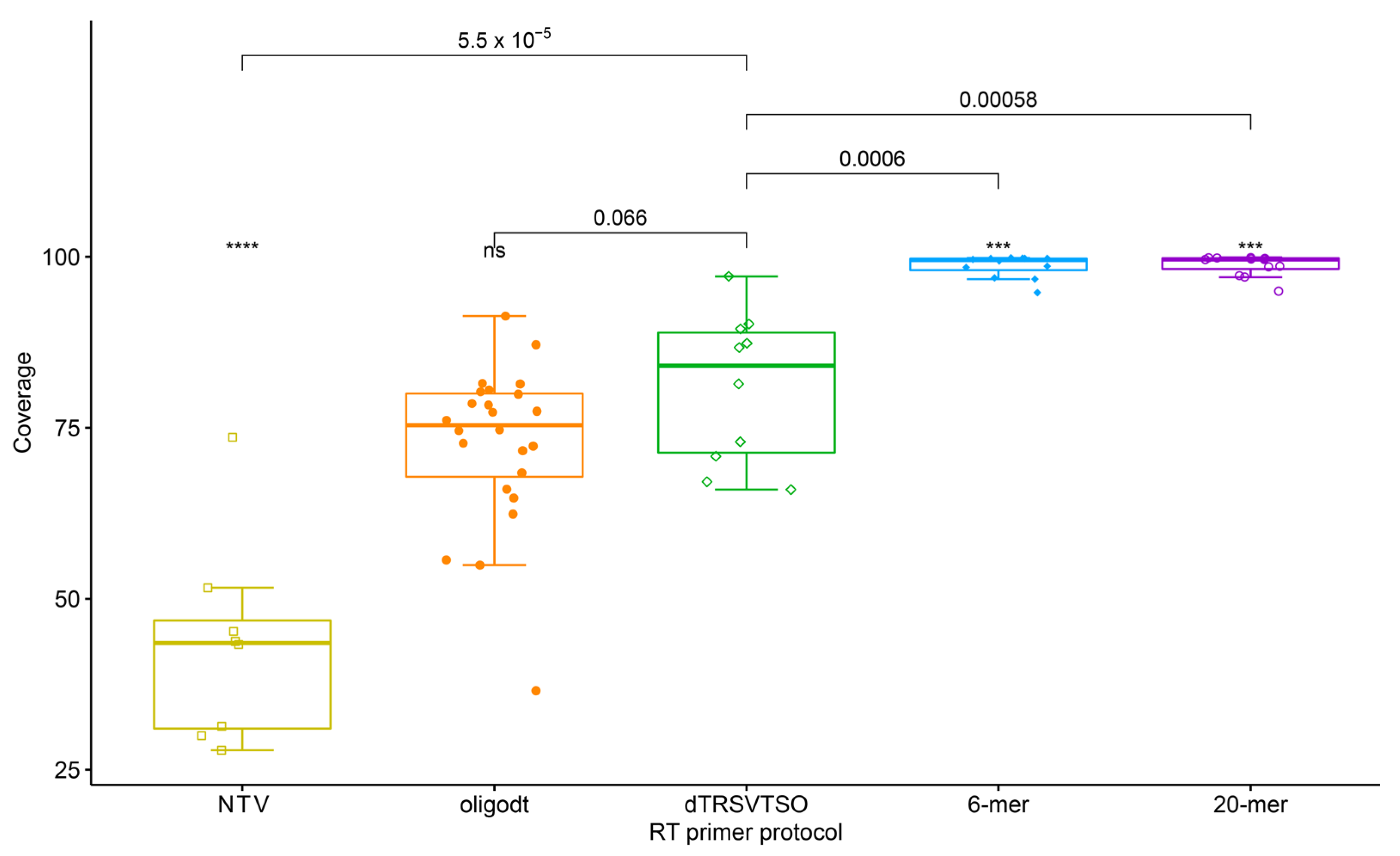
3.3. Determining Pathogen Abundance and Presence in Sequencing Library
3.4. Detection of TRSV and GLRaV-3
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Maree, H.J.; Fox, A.; Al Rwahnih, M.; Boonham, N.; Candresse, T. Application of HTS for Routine Plant Virus Diagnostics: State of the Art and Challenges. Front. Plant Sci. 2018, 9, 1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massart, S.; Chiumenti, M.; De Jonghe, K.; Glover, R.; Haegeman, A.; Koloniuk, I.; Kominek, P.; Kreuze, J.; Kutnjak, D.; Lotos, L.; et al. Virus Detection by High-Throughput Sequencing of Small RNAs: Large-Scale Performance Testing of Sequence Analysis Strategies. Phytopathology 2019, 109, 488–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malapi-Wight, M.; Kumar, L.; Mollov, D.S.; Foster, J. Implementation of next generation sequencing for high-throughput pathogen detection in sugarcane introductions grown in quarantine. Proc.Sugar J. 2017, 80, 22–23. [Google Scholar]
- Wetterstrand, K.A. DNA Sequencing Costs: Data from the NHGRI Genome Sequencing Program (GSP). Available online: https://www.genome.gov/27541954/dna-sequencing-costs-data/ (accessed on 1 May 2021).
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peserico, A.; Marcacci, M.; Malatesta, D.; Di Domenico, M.; Pratelli, A.; Mangone, I.; D’Alterio, N.; Pizzurro, F.; Cirone, F.; Zaccaria, G.; et al. Diagnosis and characterization of canine distemper virus through sequencing by MinION nanopore technology. Sci. Rep. 2019, 9, 1714. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Adam, L.; Chapados, J.; Soliman, A.; Daayf, F.; Tambong, J.T. MinION Nanopore-based detection of Clavibacter nebraskensis, the corn Goss’s wilt pathogen, and bacteriomic profiling of necrotic lesions of naturally-infected leaf samples. PLoS ONE 2021, 16, e0245333. [Google Scholar] [CrossRef]
- Honjo, M.N.; Emura, N.; Kawagoe, T.; Sugisaka, J.; Kamitani, M.; Nagano, A.J.; Kudoh, H. Seasonality of interactions between a plant virus and its host during persistent infection in a natural environment. ISME J. 2020, 14, 506–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roossinck, M.J.; Martin, D.P.; Roumagnac, P. Plant Virus Metagenomics: Advances in Virus Discovery. Phytopathology 2015, 105, 716–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.; Machleder, E.; Chenchik, A.; Li, R.; Siebert, P. Reverse Transcriptase Template Switching: A SMARTTM Approach for Full-Length cDNA Library Construction. Biotechniques 2001, 30, 892–897. [Google Scholar] [CrossRef] [Green Version]
- Ramsköld, D.; Luo, S.; Wang, Y.-C.; Li, R.; Deng, Q.; Faridani, O.R.; Daniels, G.A.; Khrebtukova, I.; Loring, J.F.; Laurent, L.C.; et al. Full-length mRNA-Seq from single-cell levels of RNA and individual circulating tumor cells. Nat. Biotechnol. 2012, 30, 777–782. [Google Scholar] [CrossRef] [Green Version]
- Turchinovich, A.; Surowy, H.; Serva, A.; Zapatka, M.; Lichter, P.; Burwinkel, B. Capture and Amplification by Tailing and Switching (CATS). An ultrasensitive ligation-independent method for generation of DNA libraries for deep sequencing from picogram amounts of DNA and RNA. RNA Biol. 2014, 11, 817–828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picelli, S.; Faridani, O.R.; Björklund, A.K.; Winberg, G.; Sagasser, S.; Sandberg, R. Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 2014, 9, 171–181. [Google Scholar] [CrossRef]
- Harbers, M.; Kato, S.; de Hoon, M.; Hayashizaki, Y.; Carninci, P.; Plessy, C. Comparison of RNA- or LNA-hybrid oligonucleotides in template-switching reactions for high-speed sequencing library preparation. BMC Genomics 2013, 14, 665. [Google Scholar] [CrossRef] [Green Version]
- Espindola, A.S.; Cardwell, K.F. Microbe Finder (MiFi®): Implementation of an Interactive Pathogen Detection Tool in Metagenomic Sequence Data. Plants 2021, 10, 250. [Google Scholar] [CrossRef] [PubMed]
- Stobbe, A.H.; Daniels, J.; Espindola, A.S.; Verma, R.; Melcher, U.; Ochoa-Corona, F.; Garzon, C.; Fletcher, J.; Schneider, W. E-probe Diagnostic Nucleic acid Analysis (EDNA): A theoretical approach for handling of next generation sequencing data for diagnostics. J. Microbiol. Methods 2013, 94, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Espindola, A.; Schneider, W.; Hoyt, P.R.; Marek, S.M.; Garzon, C. A new approach for detecting fungal and oomycete plant pathogens in next generation sequencing metagenome data utilising electronic probes. Int. J. Data Min. Bioinform. 2015, 12, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71-4–W74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arif, M.; Ochoa-Corona, F.M. Comparative assessment of 5’ A/T-rich overhang sequences with optimal and sub-optimal primers to increase PCR yields and sensitivity. Mol. Biotechnol. 2013, 55, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambino, G. Multiplex RT-PCR Method for the Simultaneous Detection of Nine Grapevine Viruses. Methods Mol. Biol. 2015, 1236, 39–47. [Google Scholar] [CrossRef]
- Converse, R.H. Occurrence of Tomato and Tobacco Ringspot Viruses and of Dagger and Other Nematodes Associated with Cultivated Highbush Blueberries in Oregon. Plant Dis. 1982, 66, 710. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Espindola, A.S.; Schneider, W.; Cardwell, K.F.; Carrillo, Y.; Hoyt, P.R.; Marek, S.M.; Melouk, H.A.; Garzon, C.D. Inferring the presence of aflatoxin-producing Aspergillus flavus strains using RNA sequencing and electronic probes as a transcriptomic screening tool. PLoS ONE 2018, 13, e0198575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zuniga, L.P.; Espindola, A.; Melouk, H.A.; Ali, A.; Cardwell, K.F.; Corona, F.O. Detection of cucurbit viruses in Oklahoma combining EDNA with Multiplex RT-PCR coupled with High Resolution Melting. In Proceedings of the 2017 APS Annual Meeting, APSNET, San Antonio, TX, USA, 5–9 August 2017; Volume 107, p. S5.36. [Google Scholar]
- Zuniga, L.P.; Daniels, J.; Espindola, A.; Corona, F.O. EDNA-Water, using deep sequencing and bioinformatics approach for water-borne plant virus detection. In Proceedings of the International Congress of Plant Pathology (ICPP), APSNET, St. Paul, MN, USA, 29 July–3 August 2018; Volume 108. [Google Scholar]
- Espindola, A.; Freire-Zapata, V.; Watanabe, L.F.M.; Corona, F.O.; Cardwell, K.F. Detecting viruses and bacteria of grapevine with Microbe Finder (MiFi) and an Oxford Nanopore sequencer. In Proceedings of the Plant Health, APS Annual Meeting; APSNET, Cleveland, OH, USA, 3–7 August 2019; Volume 109, p. S2.138. [Google Scholar]
- Espindola, A.S.; Stobbe, A.H.; Daniels, J.; Fletcher, J.; Garzon, C.D.; Schneider, W.L. Design and validation of queries for the detection of Phytophthora ramorum in simulated metagenomes. Phytopathology 2011, 101, S50. [Google Scholar]
- Espindola, A. Massively Parallel Sequencing (Mps) as a Diagnostic and Forensic Analysis Tool for Important Fungi and Chromista Plant Pathogens. Ph.D. Thesis, Oklahoma State University, Oklahoma, OK, USA, 2013. [Google Scholar]
- Bocsanczy, A.M.; Espindola, A.; Norman, D.J.; Cardwell, K.F. E-probes development for rapid, sensitive and specific pathogen detection in blueberries. In Proceedings of the ICPP Boston 2018, Plant Health in a Global Economy, APSNET, Boston, MA, USA, 29 July–3 August 2018; Volume 108, p. S1.301. [Google Scholar]
- Proano, F.; Espindola, A.; Garzon, C. Detection of multiple oomycetes in metagenomic data by Using E-probe Detection of Nucleic Analysis (EDNA). In Proceedings of the ICPP Boston 2018, Plant Health in a Global Economy, APSNET, Boston, MA, USA, 29 July–3 August 2018; Volume 108, p. S1.286. [Google Scholar]
- Espindola, A.; Roy, A.; Mavrodieva, V.A.; Cardwell, K.F. E-probe: A new diagnostic tool for detection of Dichorhaviruses associated with Citrus leprosis syndrome. In Proceedings of the Plant Health, APS Annual Meeting; APSNET, Cleveland, OH, USA, 3–7 August 2019; Volume 109, p. S2.138. [Google Scholar]
- Espindola, A.; Cardwell, K.F. Microbe Finder (MiFi): Pathogen detection in metagenomic sequence data. In Proceedings of the Plant Health, APS Annual Meeting; APSNET, Cleveland, OH, USA, 3–7 August 2019; Volume 109, p. S2.136. [Google Scholar]
- Liu, H.E.; Triboulet, M.; Zia, A.; Vuppalapaty, M.; Kidess-Sigal, E.; Coller, J.; Natu, V.S.; Shokoohi, V.; Che, J.; Renier, C.; et al. Workflow optimization of whole genome amplification and targeted panel sequencing for CTC mutation detection. NPJ Genom. Med. 2017, 2, 34. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qian, P.Y. Conservative Fragments in Bacterial 16S rRNA Genes and Primer Design for 16S Ribosomal DNA Amplicons in Metagenomic Studies. PLoS ONE 2009, 4, e7401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fierer, N.; Breitbart, M.; Nulton, J.; Salamon, P.; Lozupone, C.; Jones, R.; Robeson, M.; Edwards, R.A.; Felts, B.; Rayhawk, S.; et al. Metagenomic and Small-Subunit rRNA Analyses Reveal the Genetic Diversity of Bacteria, Archaea, Fungi, and Viruses in Soil. Appl. Environ. Microbiol. 2007, 73, 7059–7066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, C.; Li, X.; Zheng, J. Transcriptome profiling using Illumina- and SMRT-based RNA-seq of hot pepper for in-depth understanding of genes involved in CMV infection. Gene 2018, 666, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Dai, C.; Hu, C.; Liu, Z.; Kang, C. Global identification of alternative splicing via comparative analysis of SMRT- and Illumina-based RNA-seq in strawberry. Plant J. 2017, 90, 164–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Liu, C.; He, M.; Xiang, Z.; Yin, Y.; Liu, S.; Zhuang, Z. A full-length transcriptome of Sepia esculenta using a combination of single-molecule long-read (SMRT) and Illumina sequencing. Mar. Genom. 2019, 43, 54–57. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Primer Name | Type | Primer Sequence (5′ to 3′) |
|---|---|---|
| dTRSVTSO | RT primer RNA1 | AAGCAGTGGTATCAACGCAGAGTACTTAACTAGAGATTTTACTV |
| dTRSVTSO2 | RT primer RNA2 | AAGCAGTGGTATCAACGCAGAGTACCTTTAGAAAACYCAAYAGAV |
| TSO-GLRaV-3-a | RT primer | AAGCAGTGGTATCAACGCAGAGTACGACCTAACTTATTGTCGATA |
| TSO | TSO | GCTAATCATTGCAAGCAGTGGTATCAACGCAGAGTACATrGrGrG |
| TSO-PCR Primer | Amplification primer | AAGCAGTGGTATCAACGCAGAGT |
| GLRaV-3A-F | qPCR | TACGTTAAGGACGGGACACAGG |
| GLRaV-3A-R | qPCR | TGCGGCATTAATCTTCATTG |
| TRSV1-F | qPCR | CAGGGGCGTGAGTGGGGGCTC |
| TRSV1-R | qPCR | CAATACGGTAAGTGCACACCCCG |
| Sample | Ct Value (qPCR) | Mapped Reads (Ref. Genome) | Coverage (Ref. Genome) | Sequence Depth (Mean) | Control (RT Primer) | E-Probes Detection (p-Value) | ds-cDNA Protocol |
|---|---|---|---|---|---|---|---|
| GLRaV-3-1-47-1t | 18.52 | 349,668 (35.88%) | 100 | 30,498.20 | 199 | Positive (6.23 × 10−5) | TASPERT |
| GLRaV-3-1-47-2t | 21.98 | 201,241 (64.79%) | 87.53 | 14,792.60 | 129 | Positive (2.63 × 10−2) | TASPERT |
| GLRaV-3-1-47-1n | 11.21 | 3531 (13.63%) | 98.07 | 207.88 | 4,607 | Negative (>0.05) | OligodT |
| GLRaV-3-1-47-2n | 10.96 | 854 (7.24%) | 72.66 | 48.07 | 1,214 | Negative (>0.05) | OligodT |
| GLRaV-3-1-47-1t | 12.18 | 210,741 (29.13%) | 100 | 16,192.60 | 4,649 | Positive (8.24 × 10−4) | TASPERT |
| GLRaV-3-1-47-2t | 11.1 | 28,345 (35.88%) | 97.15 | 1449.09 | 2,788 | Positive (3.36 × 10−2) | TASPERT |
| GLRaV-3-1-47-3t | 12.23 | 30,005 (38.96%) | 84.92 | 1243.79 | 3205 | Negative (0.053) | TASPERT |
| TRSV-2-55-1a | 19.94 | 3287 (1.03%) | 71.64; 54.92 | 239.6; 26.03 | 249 | Positive (0.021) | OligodT |
| TRSV-2-55-2a | 20.05 | 7327 (0.90) | 81.39; 62.38 | 548; 50.46 | 618 | Positive (0.018) | OligodT |
| TRSV-2-55-3a | 19.86 | 3570 (1.32%) | 74.59; 78.54 | 269.54; 25.32 | 282 | Positive (0.038) | OligodT |
| TRSV-2-56-1a | 16.49 | 2905 (0.71%) | 78.32; 66.02 | 208.49; 31.89 | 306 | Negative (0.12) | OligodT |
| TRSV-2-56-2a | 22.11 | 3268 (0.69%) | 68.42; 72.31 | 246.77; 31.04 | 375 | Positive (0.013) | OligodT |
| TRSV-2-56-3a | 21.21 | 1042 (0.68%) | 55.68; 36.55 | 70.82; 12.04 | 143 | Negative (0.51) | OligodT |
| TRSV-2-55-1b | 14.67 | 30,075 (16.98%) | 81.41; 65.97 | 787.94; 1551.7 | 21,210 | Positive (0.032) | TASPERT |
| TRSV-2-55-3b | 15.36 | 89,605 (14.34%) | 89.45; 72.94 | 2330.65; 4459.01 | 86,618 | Positive (0.00052) | TASPERT |
| TRSV-2-56-1b | 15.05 | 190,058 (14.50%) | 97.13; 86.74 | 5418.14; 10483.7 | 186,645 | Positive (8.17 × 10−5) | TASPERT |
| TRSV-2-56-2b | 14.46 | 28,853 (10.09%) | 87.36; 70.83 | 835.93; 1552.79 | 39,209 | Positive (5.03 × 10−3) | TASPERT |
| TRSV-2-56-3b | 13.58 | 62,481 (12.15%) | 90.17; 67.12 | 1789.45; 3305.49 | 66,044 | Positive (3.26 × 10−3) | TASPERT |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Espindola, A.S.; Sempertegui-Bayas, D.; Bravo-Padilla, D.F.; Freire-Zapata, V.; Ochoa-Corona, F.; Cardwell, K.F. TASPERT: Target-Specific Reverse Transcript Pools to Improve HTS Plant Virus Diagnostics. Viruses 2021, 13, 1223. https://doi.org/10.3390/v13071223
Espindola AS, Sempertegui-Bayas D, Bravo-Padilla DF, Freire-Zapata V, Ochoa-Corona F, Cardwell KF. TASPERT: Target-Specific Reverse Transcript Pools to Improve HTS Plant Virus Diagnostics. Viruses. 2021; 13(7):1223. https://doi.org/10.3390/v13071223
Chicago/Turabian StyleEspindola, Andres S., Daniela Sempertegui-Bayas, Danny F. Bravo-Padilla, Viviana Freire-Zapata, Francisco Ochoa-Corona, and Kitty F. Cardwell. 2021. "TASPERT: Target-Specific Reverse Transcript Pools to Improve HTS Plant Virus Diagnostics" Viruses 13, no. 7: 1223. https://doi.org/10.3390/v13071223
APA StyleEspindola, A. S., Sempertegui-Bayas, D., Bravo-Padilla, D. F., Freire-Zapata, V., Ochoa-Corona, F., & Cardwell, K. F. (2021). TASPERT: Target-Specific Reverse Transcript Pools to Improve HTS Plant Virus Diagnostics. Viruses, 13(7), 1223. https://doi.org/10.3390/v13071223