
Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. HSV-1 and -2 Infection
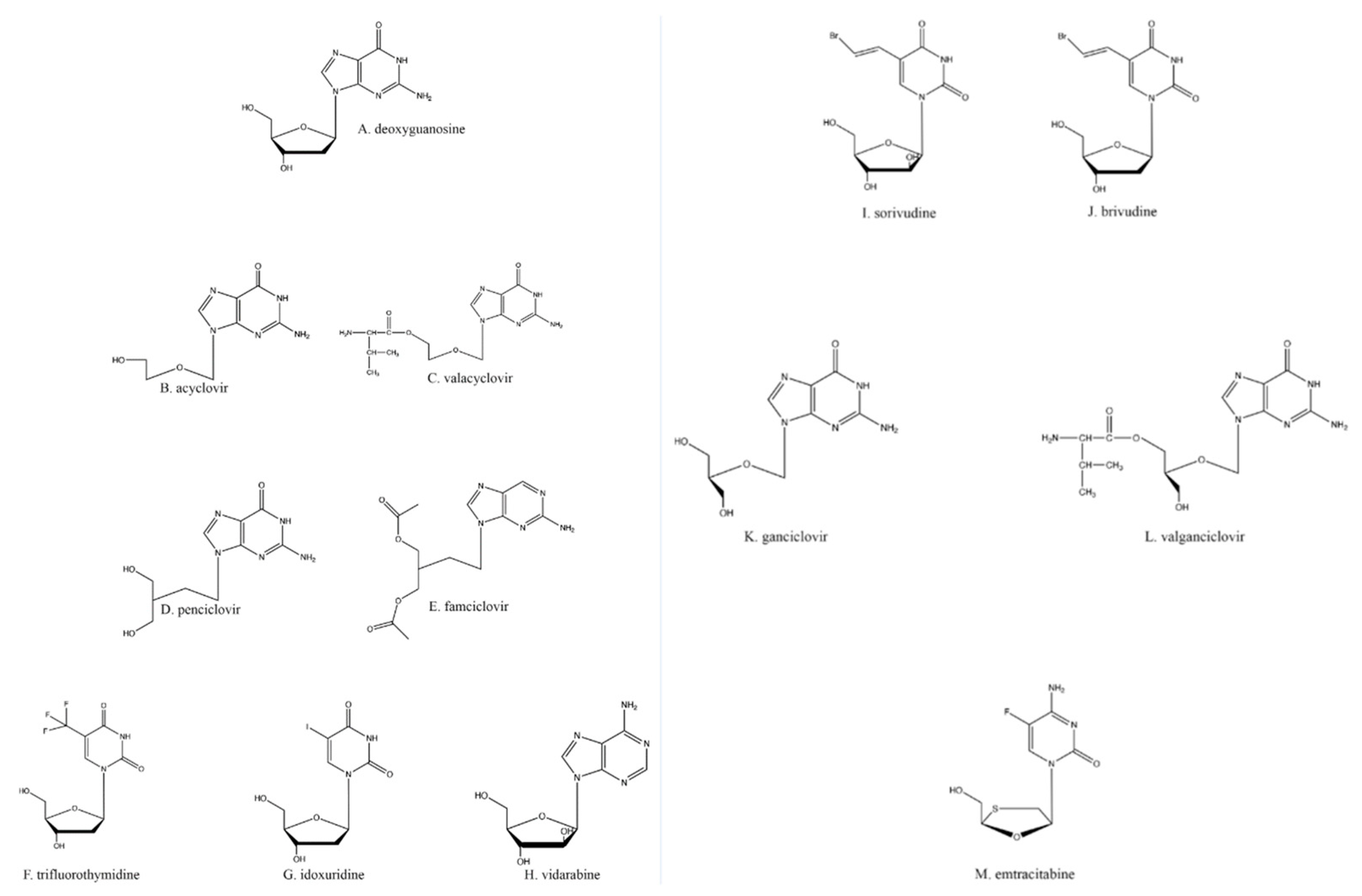
3. Nucleoside Analogs
4. Acyclovir, the First in Its Class of Antiherpetic Drugs
5. Other Nucleoside Analogs
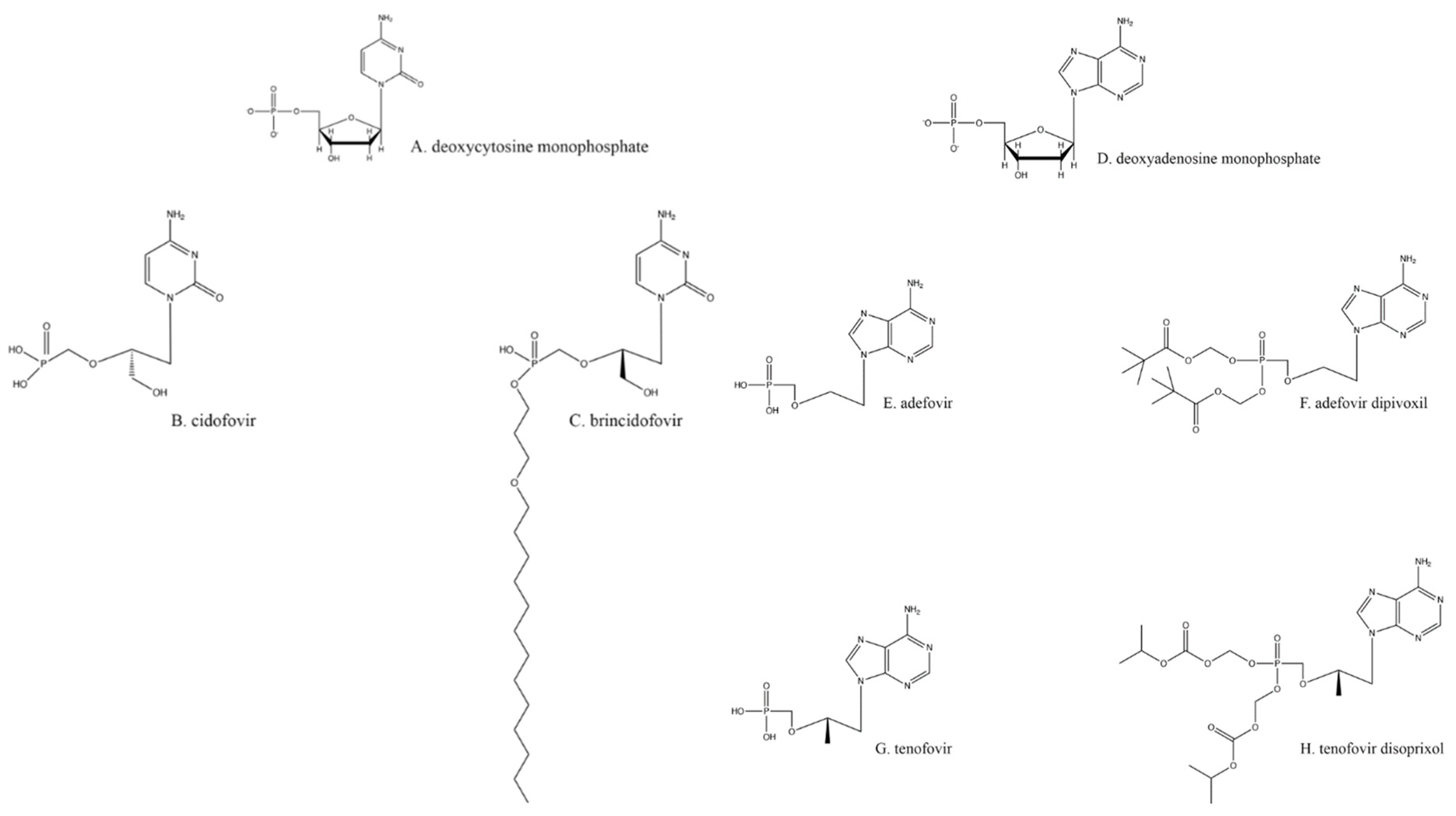
6. Nucleotide Analogs: Cidofovir, Adefovir, and Brincidofovir
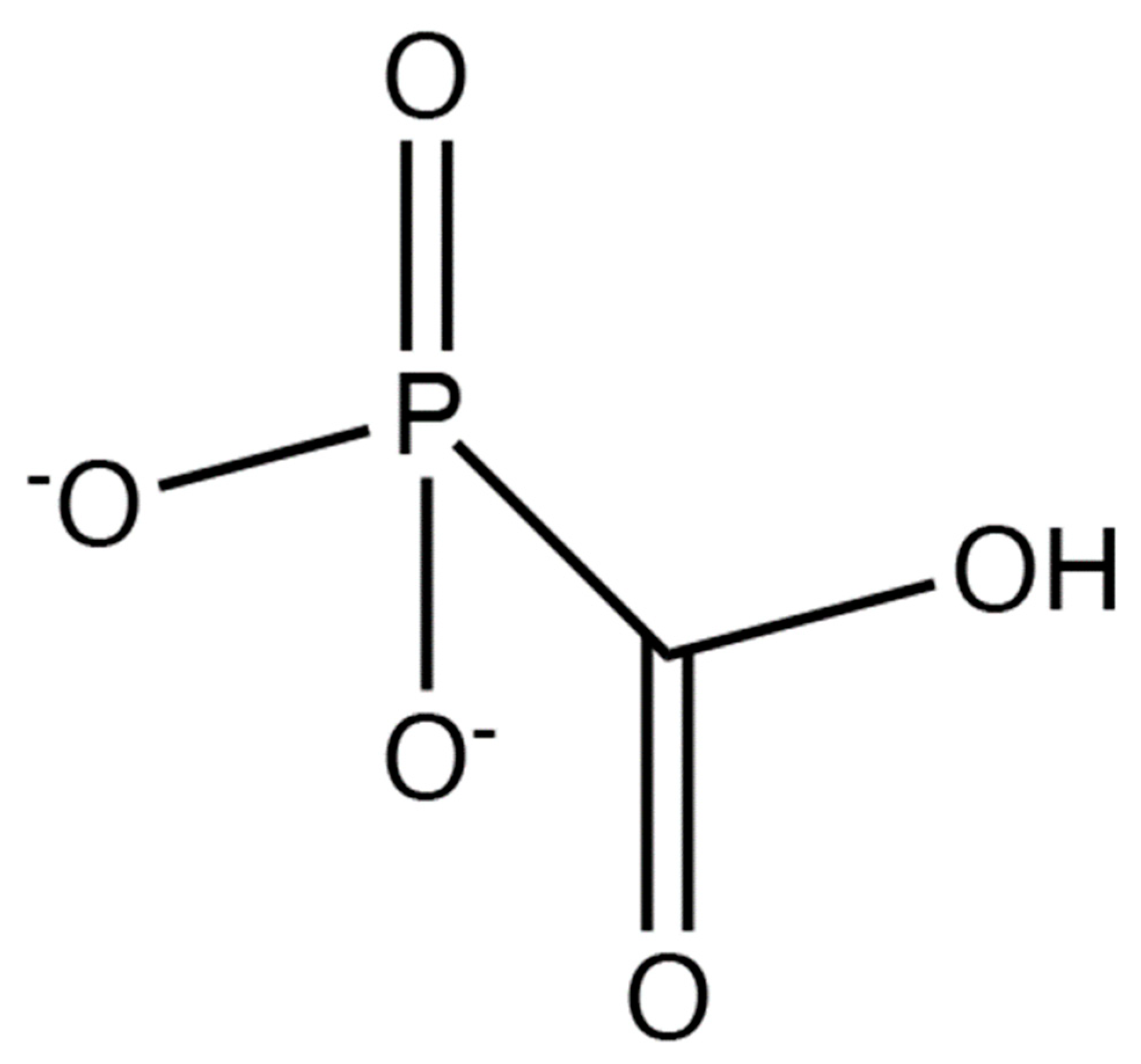
7. Non-Nucleoside/Nucleotide Inhibition of Herpes DNA Polymerase
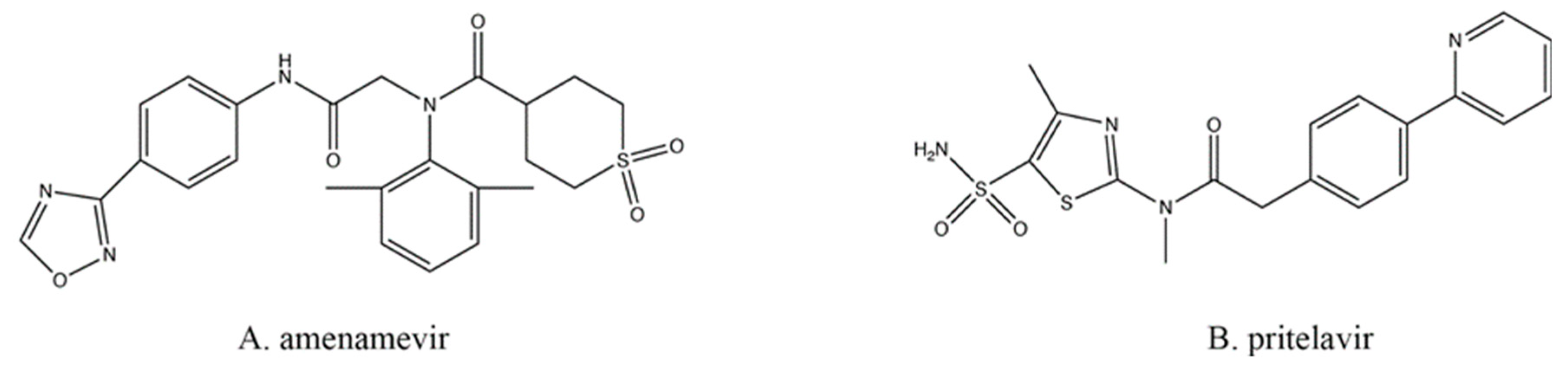
8. Helicase/Primase Inhibitors
9. Binding and Entry Inhibition
10. Licensed Drugs for Other Infectious Agents Can Also Inhibit Herpesviruses
11. Summary
Author Contributions
Funding
Conflicts of Interest
References
- Kaufman, H.; Martola, E.L.; Dohlman, C. Use of 5-iodo-2′-deoxyuridine (IDU) in treatment of herpes simplex keratitis. Arch. Ophthalmol. 1962, 68, 235–239. [Google Scholar] [CrossRef]
- Kaufman, H.E. Clinical cure of herpes simplex keratitis by 5-iodo-2-deoxyuridine. Proc. Soc. Exp. Biol. Med. 1962, 109, 251–252. [Google Scholar] [CrossRef] [PubMed]
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes Simplex Viruses. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Cohen, J.I., Griffin, D.E., Lamb, R.A., Martin, M.A., Racaniello, V.R., Roizman, B., Eds.; Wolters Kluwer/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; pp. 1823–1897. [Google Scholar]
- Looker, K.J.; Garnett, G.P. A systematic review of the epidemiology and interaction of herpes simplex virus types 1 and 2. Sex. Transm. Infect. 2005, 81, 103–107. [Google Scholar] [CrossRef]
- Kalu, N.N.; Desai, P.J.; Shirley, C.M.; Gibson, W.; Dennis, P.A.; Ambinder, R.F. Nelfinavir inhibits maturation and export of herpes simplex virus 1. J. Virol. 2014, 88, 5455–5461. [Google Scholar] [CrossRef] [Green Version]
- Jia, X.; Schols, D.; Meier, C. Lipophilic Triphosphate Prodrugs of Various Nucleoside Analogues. J. Med. Chem. 2020, 63, 6991–7007. [Google Scholar] [CrossRef]
- Berdis, A.J. DNA polymerases as therapeutic targets. Biochemistry 2008, 47, 8253–8260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elion, G.B. Mechanism of action and selectivity of acyclovir. Am. J. Med. 1982, 73, 7–13. [Google Scholar] [CrossRef]
- Furman, P.A.; St Clair, M.H.; Spector, T. Acyclovir triphosphate is a suicide inactivator of the herpes simplex virus DNA polymerase. J. Biol. Chem. 1984, 259, 9575–9579. [Google Scholar] [CrossRef]
- Yajima, M.; Yamada, H.; Takemoto, M.; Daikoku, T.; Yoshida, Y.; Long, T.; Okuda, T.; Shiraki, K. Profile of anti-herpetic action of ASP2151 (amenamevir) as a helicase-primase inhibitor. Antivir. Res. 2017, 139, 95–101. [Google Scholar] [CrossRef]
- Xiong, X.; Smith, J.L.; Chen, M.S. Effect of incorporation of cidofovir into DNA by human cytomegalovirus DNA polymerase on DNA elongation. Antimicrob. Agents Chemother. 1997, 41, 594–599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunha, A.C.; Ferreira, V.F.; Vaz, M.G.F.; Cassaro, R.A.A.; Resende, J.; Sacramento, C.Q.; Costa, J.; Abrantes, J.L.; Souza, T.M.L.; Jordao, A.K. Chemistry and anti-herpes simplex virus type 1 evaluation of 4-substituted-1H-1,2,3-triazole-nitroxyl-linked hybrids. Mol. Divers. 2020. [Google Scholar] [CrossRef]
- McGuirt, P.V.; Furman, P.A. Acyclovir inhibition of viral DNA chain elongation in herpes simplex virus-infected cells. Am. J. Med. 1982, 73, 67–71. [Google Scholar] [CrossRef]
- Topalis, D.; Gillemot, S.; Snoeck, R.; Andrei, G. Distribution and effects of amino acid changes in drug-resistant alpha and beta herpesviruses DNA polymerase. Nucleic Acids Res. 2016, 44, 9530–9554. [Google Scholar] [PubMed] [Green Version]
- Crumpacker, C.S.; Schnipper, L.E.; Chartrand, P.; Knopf, K.W. Genetic mechanisms of resistance to acyclovir in herpes simplex virus. Am. J. Med. 1982, 73, 361–368. [Google Scholar] [CrossRef]
- Van Dyke, R.B.; Connor, J.D.; Wyborny, C.; Hintz, M.; Keeney, R.E. Pharmacokinetics of orally administered acyclovir in patients with herpes progenitalis. Am. J. Med. 1982, 73, 172–175. [Google Scholar] [CrossRef]
- Gurgel Assis, M.S.; Fernandes Pedrosa, T.C.; de Moraes, F.S.; Caldeira, T.G.; Pereira, G.R.; de Souza, J.; Ruela, A.L.M. Novel Insights to Enhance Therapeutics with Acyclovir in the Management of Herpes Simplex Encephalitis. J. Pharm. Sci. 2021, 110, 1557–1571. [Google Scholar] [CrossRef] [PubMed]
- Birkmann, A.; Zimmermann, H. HSV antivirals-current and future treatment options. Curr. Opin. Virol. 2016, 18, 9–13. [Google Scholar] [CrossRef] [PubMed]
- Lai, L.; Xu, Z.; Zhou, J.; Lee, K.D.; Amidon, G.L. Molecular basis of prodrug activation by human valacyclovirase, an alpha-amino acid ester hydrolase. J. Biol. Chem. 2008, 283, 9318–9327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marsillach, J.; Suzuki, S.M.; Richter, R.J.; McDonald, M.G.; Rademacher, P.M.; MacCoss, M.J.; Hsieh, E.J.; Rettie, A.E.; Furlong, C.E. Human valacyclovir hydrolase/biphenyl hydrolase-like protein is a highly efficient homocysteine thiolactonase. PLoS ONE 2014, 9, e110054. [Google Scholar] [CrossRef] [PubMed]
- Earnshaw, D.L.; Bacon, T.H.; Darlison, S.J.; Edmonds, K.; Perkins, R.M.; Vere Hodge, R.A. Mode of antiviral action of penciclovir in MRC-5 cells infected with herpes simplex virus type 1 (HSV-1), HSV-2, and varicella-zoster virus. Antimicrob. Agents Chemother. 1992, 36, 2747–2757. [Google Scholar] [CrossRef] [Green Version]
- Boyd, M.R.; Bacon, T.H.; Sutton, D. Antiherpesvirus activity of 9-(4-hydroxy-3-hydroxymethylbut-1-yl) guanine (BRL 39123) in animals. Antimicrob. Agents Chemother. 1988, 32, 358–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gill, K.S.; Wood, M.J. The clinical pharmacokinetics of famciclovir. Clin. Pharm. 1996, 31, 1–8. [Google Scholar] [CrossRef]
- Vere Hodge, R.A.; Sutton, D.; Boyd, M.R.; Harnden, M.R.; Jarvest, R.L. Selection of an oral prodrug (BRL 42810; famciclovir) for the antiherpesvirus agent BRL 39123 [9-(4-hydroxy-3-hydroxymethylbut-l-yl)guanine; penciclovir]. Antimicrob. Agents Chemother. 1989, 33, 1765–1773. [Google Scholar] [CrossRef] [Green Version]
- Filer, C.W.; Allen, G.D.; Brown, T.A.; Fowles, S.E.; Hollis, F.J.; Mort, E.E.; Prince, W.T.; Ramji, J.V. Metabolic and pharmacokinetic studies following oral administration of 14C-famciclovir to healthy subjects. Xenobiotica 1994, 24, 357–368. [Google Scholar] [CrossRef]
- Clarke, S.E.; Harrell, A.W.; Chenery, R.J. Role of aldehyde oxidase in the in vitro conversion of famciclovir to penciclovir in human liver. Drug Metab. Dispos. 1995, 23, 251–254. [Google Scholar]
- Agrahari, V.; Mandal, A.; Agrahari, V.; Trinh, H.M.; Joseph, M.; Ray, A.; Hadji, H.; Mitra, R.; Pal, D.; Mitra, A.K. A comprehensive insight on ocular pharmacokinetics. Drug Deliv. Transl. Res. 2016, 6, 735–754. [Google Scholar] [CrossRef]
- Silvestri, D.L.; Corey, L.; Holmes, K.K. Ineffectiveness of topical idoxuridine in dimethyl sulfoxide for therapy for genital herpes. JAMA 1982, 248, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, F.; Komoto, I.; Oka, H.; Kuwata, K.; Takeuchi, M.; Nakagawa, F.; Yoshisue, K.; Chiba, M. Exposure-dependent incorporation of trifluridine into DNA of tumors and white blood cells in tumor-bearing mouse. Cancer Chemother. Pharm. 2015, 76, 325–333. [Google Scholar] [CrossRef]
- Sharma, S.; Mehndiratta, S.; Kumar, S.; Singh, J.; Bedi, P.M.; Nepali, K. Purine Analogues as Kinase Inhibitors: A Review. Recent Pat. Anticancer Drug Discov. 2015, 10, 308–341. [Google Scholar] [CrossRef] [PubMed]
- Whitley, R.J.; Tucker, B.C.; Kinkel, A.W.; Barton, N.H.; Pass, R.F.; Whelchel, J.D.; Cobbs, C.G.; Diethelm, A.G.; Buchanan, R.A. Pharmacology, tolerance, and antiviral activity of vidarabine monophosphate in humans. Antimicrob. Agents Chemother. 1980, 18, 709–715. [Google Scholar] [CrossRef] [Green Version]
- Aebersold, P.M. Relative mutagenicity of nucleoside virostatic drugs in Chinese hamster ovary cells. Adv. Ophthalmol. 1979, 38, 214–221. [Google Scholar]
- Seley-Radtke, K.L.; Yates, M.K. The evolution of nucleoside analogue antivirals: A review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antivir. Res. 2018, 154, 66–86. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, H.E. Antimetabolite drug therapy in herpes simplex. Ophthalmology 1980, 87, 135–139. [Google Scholar] [CrossRef]
- De Clercq, E. Discovery and development of BVDU (brivudin) as a therapeutic for the treatment of herpes zoster. Biochem. Pharmacol. 2004, 68, 2301–2315. [Google Scholar] [CrossRef]
- Wassilew, S.W.; Wutzler, P.; Brivddin Herpes Zoster Study Group. Oral brivudin in comparison with acyclovir for herpes zoster: A survey study on postherpetic neuralgia. Antivir. Res. 2003, 59, 57–60. [Google Scholar] [CrossRef]
- Andrei, G.; Balzarini, J.; Fiten, P.; De Clercq, E.; Opdenakker, G.; Snoeck, R. Characterization of herpes simplex virus type 1 thymidine kinase mutants selected under a single round of high-dose brivudin. J. Virol. 2005, 79, 5863–5869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, S.; Faulds, D. Ganciclovir. An update of its use in the prevention of cytomegalovirus infection and disease in transplant recipients. Drugs 1998, 56, 115–146. [Google Scholar] [CrossRef]
- Gilbert, C.; Bestman-Smith, J.; Boivin, G. Resistance of herpesviruses to antiviral drugs: Clinical impacts and molecular mechanisms. Drug Resist. Updates 2002, 5, 88–114. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Grill, S.P.; Dutschman, G.E.; Nakayama, K.; Bastow, K.F. Metabolism of 9-(1,3-dihydroxy-2-propoxymethyl)guanine, a new anti-herpes virus compound, in herpes simplex virus-infected cells. J. Biol. Chem. 1983, 258, 12460–12464. [Google Scholar] [CrossRef]
- Cheng, Y.C.; Huang, E.S.; Lin, J.C.; Mar, E.C.; Pagano, J.S.; Dutschman, G.E.; Grill, S.P. Unique spectrum of activity of 9-[(1,3-dihydroxy-2-propoxy)methyl]-guanine against herpesviruses in vitro and its mode of action against herpes simplex virus type 1. Proc. Natl. Acad. Sci. USA 1983, 80, 2767–2770. [Google Scholar] [CrossRef] [Green Version]
- Jung, D.; Dorr, A. Single-dose pharmacokinetics of valganciclovir in HIV- and CMV-seropositive subjects. J. Clin. Pharmacol. 1999, 39, 800–804. [Google Scholar] [CrossRef]
- Brown, F.; Banken, L.; Saywell, K.; Arum, I. Pharmacokinetics of valganciclovir and ganciclovir following multiple oral dosages of valganciclovir in HIV-and CMV-seropositive volunteers. Clin. Pharm. 1999, 37, 167–176. [Google Scholar] [CrossRef]
- Morfin, F.; Thouvenot, D. Herpes simplex virus resistance to antiviral drugs. J. Clin. Virol. 2003, 26, 29–37. [Google Scholar] [CrossRef]
- Bronson, J.J.; Ho, H.T.; De Boeck, H.; Woods, K.; Ghazzouli, I.; Martin, J.C.; Hitchcock, M.J. Biochemical pharmacology of acyclic nucleotide analogues. Ann. N. Y. Acad Sci. 1990, 616, 398–407. [Google Scholar] [CrossRef]
- Andrei, G.; Snoeck, R.; De Clercq, E.; Esnouf, R.; Fiten, P.; Opdenakker, G. Resistance of herpes simplex virus type 1 against different phosphonylmethoxyalkyl derivatives of purines and pyrimidines due to specific mutations in the viral DNA polymerase gene. J. Gen. Virol. 2000, 81, 639–648. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R. Drug-resistant cytomegalovirus: Clinical implications of specific mutations. Curr. Opin. Organ. Transpl. 2018, 23, 388–394. [Google Scholar] [CrossRef]
- Meier, P.; Dautheville-Guibal, S.; Ronco, P.M.; Rossert, J. Cidofovir-induced end-stage renal failure. Nephrol. Dial. Transpl. 2002, 17, 148–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, A. Antiviral treatment of cytomegalovirus infection. Infect. Disord. Drug Targets 2011, 11, 475–503. [Google Scholar] [CrossRef]
- Marty, F.M.; Winston, D.J.; Chemaly, R.F.; Mullane, K.M.; Shore, T.B.; Papanicolaou, G.A.; Chittick, G.; Brundage, T.M.; Wilson, C.; Morrison, M.E.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 3 Trial of Oral Brincidofovir for Cytomegalovirus Prophylaxis in Allogeneic Hematopoietic Cell Transplantation. Biol. Blood Marrow Transpl. 2019, 25, 369–381. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, P.; Lumley, S. Cytomegalovirus. Curr. Opin. Infect. Dis. 2014, 27, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J.; Naesens, L.; De Clercq, E. New antivirals-mechanism of action and resistance development. Curr. Opin. Microbiol. 1998, 1, 535–546. [Google Scholar] [CrossRef]
- Aduma, P.; Connelly, M.C.; Srinivas, R.V.; Fridland, A. Metabolic diversity and antiviral activities of acyclic nucleoside phosphonates. Mol. Pharmacol. 1995, 47, 816–822. [Google Scholar]
- Foster, S.A.; Cerny, J.; Cheng, Y.C. Herpes simplex virus-specified DNA polymerase is the target for the antiviral action of 9-(2-phosphonylmethoxyethyl)adenine. J. Biol. Chem. 1991, 266, 238–244. [Google Scholar] [CrossRef]
- Huang, C.; Yang, X.H.; Yang, Y.L.; Huang, A.L.; Shi, X.F. Clinical-features analysis on 926 patients with virological breakthrough in chronic hepatitis B receiving nucleos(t)ide analogues. Eur. J. Intern. Med. 2018, 53, e9–e10. [Google Scholar] [CrossRef] [PubMed]
- Cundy, K.C.; Barditch-Crovo, P.; Walker, R.E.; Collier, A.C.; Ebeling, D.; Toole, J.; Jaffe, H.S. Clinical pharmacokinetics of adefovir in human immunodeficiency virus type 1-infected patients. Antimicrob. Agents Chemother. 1995, 39, 2401–2405. [Google Scholar] [CrossRef] [Green Version]
- Law, S.T.; Li, K.K.; Ho, Y.Y. Acquired Fanconi syndrome associated with prolonged adefovir dipivoxil therapy in a chronic hepatitis B patient. Am. J. Ther. 2013, 20, e713–e716. [Google Scholar] [CrossRef] [PubMed]
- Vashishtha, A.K.; Kuchta, R.D. Effects of Acyclovir, Foscarnet, and Ribonucleotides on Herpes Simplex Virus-1 DNA Polymerase: Mechanistic Insights and a Novel Mechanism for Preventing Stable Incorporation of Ribonucleotides into DNA. Biochemistry 2016, 55, 1168–1177. [Google Scholar] [CrossRef] [Green Version]
- Crumpacker, C.S. Mechanism of action of foscarnet against viral polymerases. Am. J. Med. 1992, 92, S3–S7. [Google Scholar] [CrossRef]
- Leowattana, W. Antiviral Drugs and Acute Kidney Injury (AKI). Infect. Disord. Drug Targets 2019, 19, 375–382. [Google Scholar] [CrossRef]
- Mareri, A.; Lasorella, S.; Iapadre, G.; Maresca, M.; Tambucci, R.; Nigro, G. Anti-viral therapy for congenital cytomegalovirus infection: Pharmacokinetics, efficacy and side effects. J. Matern. Fetal Neonatal Med. 2016, 29, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.A.; Gambertoglio, J.G.; Aweeka, F.T.; Causey, D.M.; Portale, A.A. Foscarnet-induced hypocalcemia and effects of foscarnet on calcium metabolism. J. Clin. Endocrinol. Metab. 1991, 72, 1130–1135. [Google Scholar] [CrossRef]
- Jayaweera, D.T. Minimising the dosage-limiting toxicities of foscarnet induction therapy. Drug Saf. 1997, 16, 258–266. [Google Scholar] [CrossRef]
- Stenberg, K.; Skog, S.; Tribukait, B. Concentration-dependent effects of foscarnet on the cell cycle. Antimicrob. Agents Chemother. 1985, 28, 802–806. [Google Scholar] [CrossRef] [Green Version]
- Matthews, J.T.; Terry, B.J.; Field, A.K. The structure and function of the HSV DNA replication proteins: Defining novel antiviral targets. Antivir. Res. 1993, 20, 89–114. [Google Scholar] [CrossRef]
- Shoji, N.; Tanese, K.; Sasaki, A.; Horiuchi, T.; Utsuno, Y.; Fukuda, K.; Hoshino, Y.; Noda, S.; Minami, H.; Asakura, W.; et al. Pharmaceuticals and Medical Device Agency approval summary: Amenamevir for the treatment of herpes zoster. J. Dermatol. 2020, 47, 683–688. [Google Scholar] [CrossRef]
- Shiraki, K. Antiviral Drugs Against Alphaherpesvirus. Adv. Exp. Med. Biol. 2018, 1045, 103–122. [Google Scholar]
- Field, H.J.; Biswas, S. Antiviral drug resistance and helicase-primase inhibitors of herpes simplex virus. Drug Resist. Updates 2011, 14, 45–51. [Google Scholar] [CrossRef]
- Biswas, S.; Jennens, L.; Field, H.J. Single amino acid substitutions in the HSV-1 helicase protein that confer resistance to the helicase-primase inhibitor BAY 57-1293 are associated with increased or decreased virus growth characteristics in tissue culture. Arch. Virol. 2007, 152, 1489–1500. [Google Scholar] [CrossRef] [PubMed]
- Liuzzi, M.; Kibler, P.; Bousquet, C.; Harji, F.; Bolger, G.; Garneau, M.; Lapeyre, N.; McCollum, R.S.; Faucher, A.M.; Simoneau, B.; et al. Isolation and characterization of herpes simplex virus type 1 resistant to aminothiazolylphenyl-based inhibitors of the viral helicase-primase. Antivir. Res. 2004, 64, 161–170. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Gallay, P. Curing a viral infection by targeting the host: The example of cyclophilin inhibitors. Antivir. Res. 2013, 99, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W.; Hwang, C.B. On the mutation rate of herpes simplex virus type 1. Genetics 2005, 170, 969–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renner, D.W.; Szpara, M.L. Impacts of Genome-Wide Analyses on Our Understanding of Human Herpesvirus Diversity and Evolution. J. Virol. 2018, 92, e00908-17. [Google Scholar] [CrossRef] [Green Version]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [Green Version]
- Katz, D.H.; Marcelletti, J.F.; Khalil, M.H.; Pope, L.E.; Katz, L.R. Antiviral activity of 1-docosanol, an inhibitor of lipid-enveloped viruses including herpes simplex. Proc. Natl. Acad. Sci. USA 1991, 88, 10825–10829. [Google Scholar] [CrossRef] [Green Version]
- Pope, L.E.; Marcelletti, J.F.; Katz, L.R.; Lin, J.Y.; Katz, D.H.; Parish, M.L.; Spear, P.G. The anti-herpes simplex virus activity of n-docosanol includes inhibition of the viral entry process. Antivir. Res. 1998, 40, 85–94. [Google Scholar] [CrossRef]
- Marcelletti, J.F. Synergistic inhibition of herpesvirus replication by docosanol and antiviral nucleoside analogs. Antivir. Res. 2002, 56, 153–166. [Google Scholar] [CrossRef]
- Woo, S.B.; Challacombe, S.J. Management of recurrent oral herpes simplex infections. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endodontol. 2007, 103 (Suppl. S12), e1–e18. [Google Scholar] [CrossRef] [PubMed]
- Usatine, R.P.; Tinitigan, R. Nongenital herpes simplex virus. Am. Fam. Phys. 2010, 82, 1075–1082. [Google Scholar]
- Leung, D.T.; Sacks, S.L. Docosanol: A topical antiviral for herpes labialis. Expert Opin. Pharm. 2004, 5, 2567–2571. [Google Scholar] [CrossRef]
- Deeks, E.D. Bictegravir/Emtricitabine/Tenofovir Alafenamide: A Review in HIV-1 Infection. Drugs 2018, 78, 1817–1828. [Google Scholar] [CrossRef] [Green Version]
- Marcus, J.L.; Glidden, D.V.; McMahan, V.; Lama, J.R.; Mayer, K.H.; Liu, A.Y.; Montoya-Herrera, O.; Casapia, M.; Hoagland, B.; Grant, R.M. Daily oral emtricitabine/tenofovir preexposure prophylaxis and herpes simplex virus type 2 among men who have sex with men. PLoS ONE 2014, 9, e91513. [Google Scholar] [CrossRef]
- Celum, C.; Morrow, R.A.; Donnell, D.; Hong, T.; Hendrix, C.W.; Thomas, K.K.; Fife, K.H.; Nakku-Joloba, E.; Mujugira, A.; Baeten, J.M. Daily oral tenofovir and emtricitabine-tenofovir preexposure prophylaxis reduces herpes simplex virus type 2 acquisition among heterosexual HIV-1-uninfected men and women: A subgroup analysis of a randomized trial. Ann. Intern. Med. 2014, 161, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Figueroa, D.B.; Madeen, E.P.; Tillotson, J.; Richardson, P.; Cottle, L.; McCauley, M.; Landovitz, R.J.; Andrade, A.; Hendrix, C.W.; Mayer, K.H.; et al. Genetic Variation of the Kinases That Phosphorylate Tenofovir and Emtricitabine in Peripheral Blood Mononuclear Cells. AIDS Res. Hum. Retrovir. 2018, 34, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Baeten, J.M.; Donnell, D.; Mugo, N.R.; Ndase, P.; Thomas, K.K.; Campbell, J.D.; Wangisi, J.; Tappero, J.W.; Bukusi, E.A.; Cohen, C.R.; et al. Single-agent tenofovir versus combination emtricitabine plus tenofovir for pre-exposure prophylaxis for HIV-1 acquisition: An update of data from a randomised, double-blind, phase 3 trial. Lancet Infect. Dis. 2014, 14, 1055–1064. [Google Scholar] [CrossRef] [Green Version]
- Chaix, M.L.; Charreau, I.; Pintado, C.; Delaugerre, C.; Mahjoub, N.; Cotte, L.; Capitant, C.; Raffi, F.; Cua, E.; Pialoux, G.; et al. Effect of On-Demand Oral Pre-exposure Prophylaxis With Tenofovir/Emtricitabine on Herpes Simplex Virus-1/2 Incidence Among Men Who Have Sex With Men: A Substudy of the ANRS IPERGAY Trial. Open Forum Infect. Dis. 2018, 5, ofy295. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadowski, L.A.; Upadhyay, R.; Greeley, Z.W.; Margulies, B.J. Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2. Viruses 2021, 13, 1228. https://doi.org/10.3390/v13071228
Sadowski LA, Upadhyay R, Greeley ZW, Margulies BJ. Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2. Viruses. 2021; 13(7):1228. https://doi.org/10.3390/v13071228
Chicago/Turabian StyleSadowski, Lauren A., Rista Upadhyay, Zachary W. Greeley, and Barry J. Margulies. 2021. "Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2" Viruses 13, no. 7: 1228. https://doi.org/10.3390/v13071228
APA StyleSadowski, L. A., Upadhyay, R., Greeley, Z. W., & Margulies, B. J. (2021). Current Drugs to Treat Infections with Herpes Simplex Viruses-1 and -2. Viruses, 13(7), 1228. https://doi.org/10.3390/v13071228