
Proteome Analysis in PAM Cells Reveals That African Swine Fever Virus Can Regulate the Level of Intracellular Polyamines to Facilitate Its Own Replication through ARG1

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
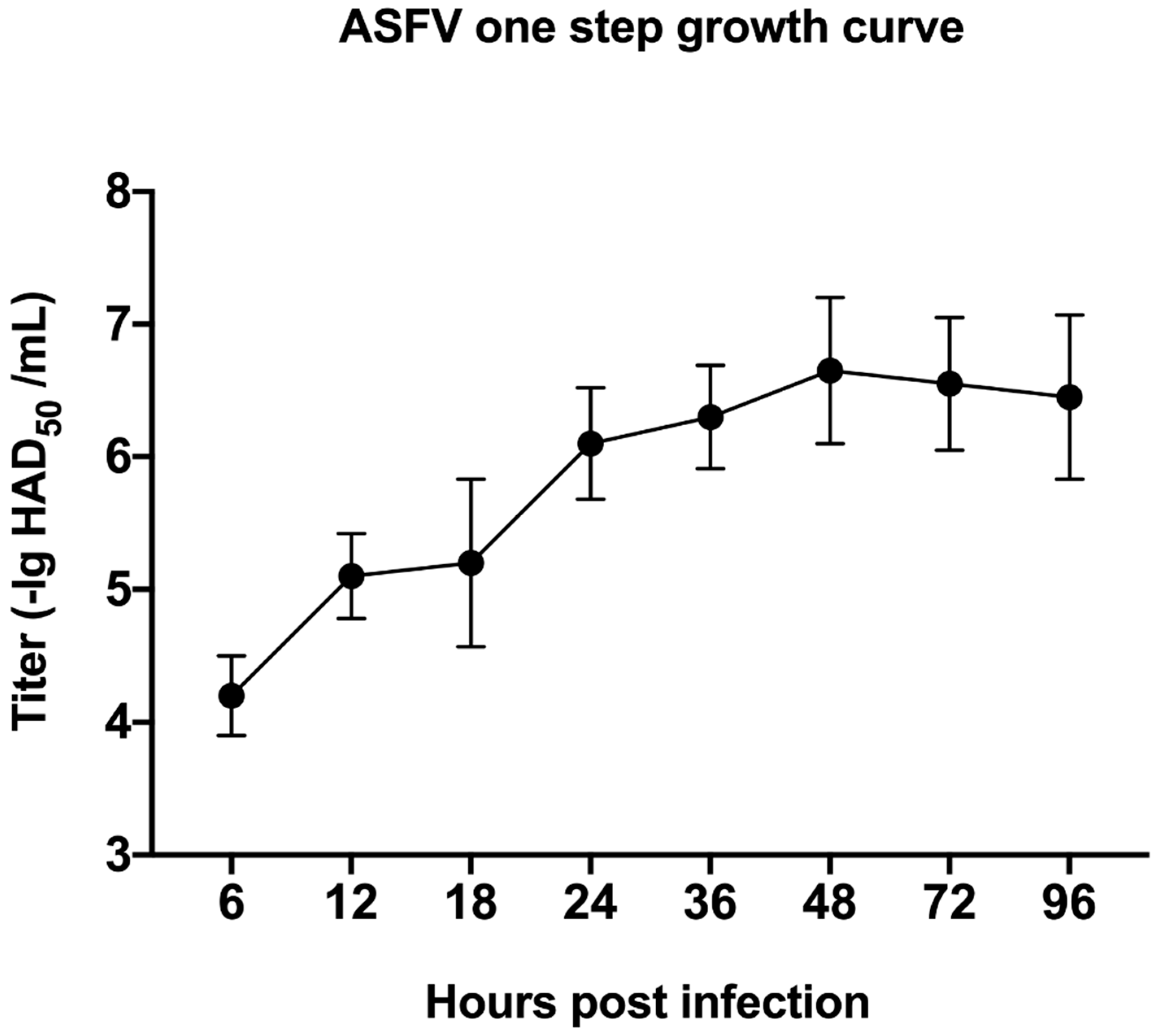
2.2. Growth Curve Analysis of ASFV
2.3. HAD50 Test Assay
2.4. Indirect Immunofluorescence Assay
2.5. Cell Sample Preparation
2.6. Protein Digestion
2.7. TMT Labeling
2.8. Mass Spectrometry
2.9. Bioinformatic Analysis
2.10. Western Blot Analysis of Candidate Proteins
2.11. Real-Time Quantitative PCR (qPCR)
2.12. Nitric Oxide Detection
2.13. Determination of Intracellular Polyamines Level
2.14. The Effect of L-Arginine on ASFV Replication
2.15. siRNA Transfection
2.16. Biosafety Statement
2.17. Statistical Analysis
3. Results
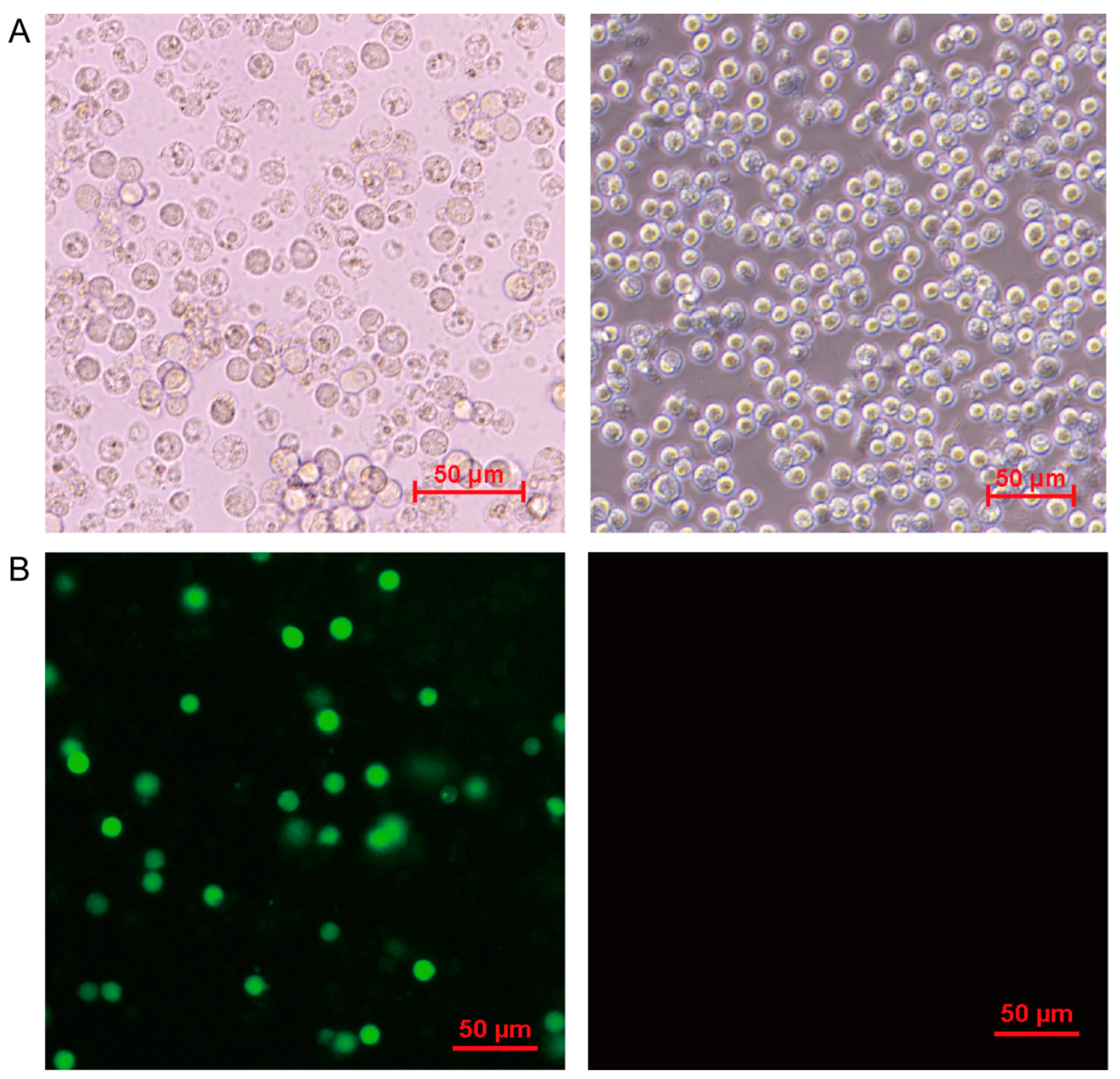
3.1. Kinetics of ASFV-Induced Cytopathology in PAM Cells
3.2. Cellular Protein Identification and Quantification by TMT Method
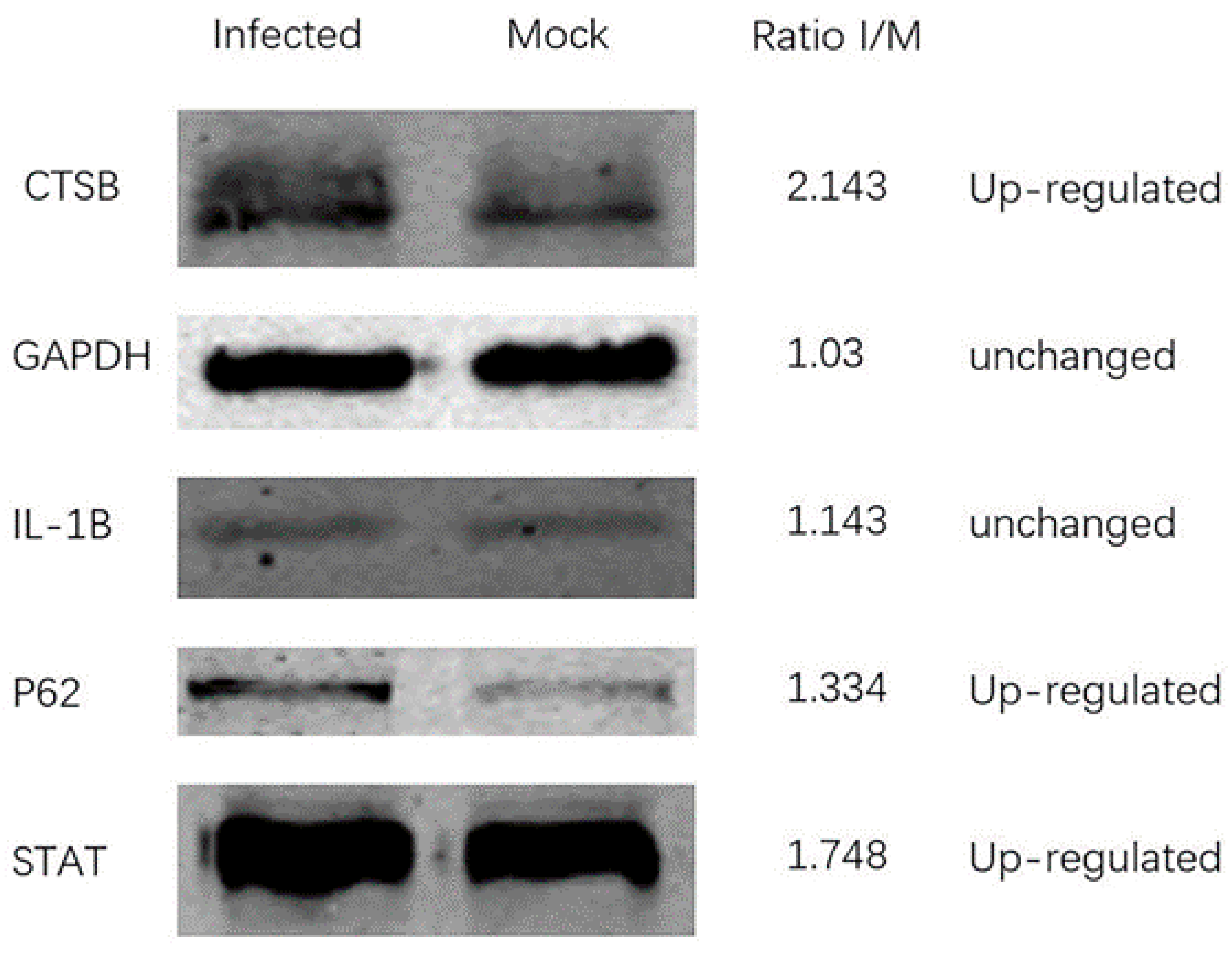
3.3. Verify Protein Changes in TMT Proteomics Data
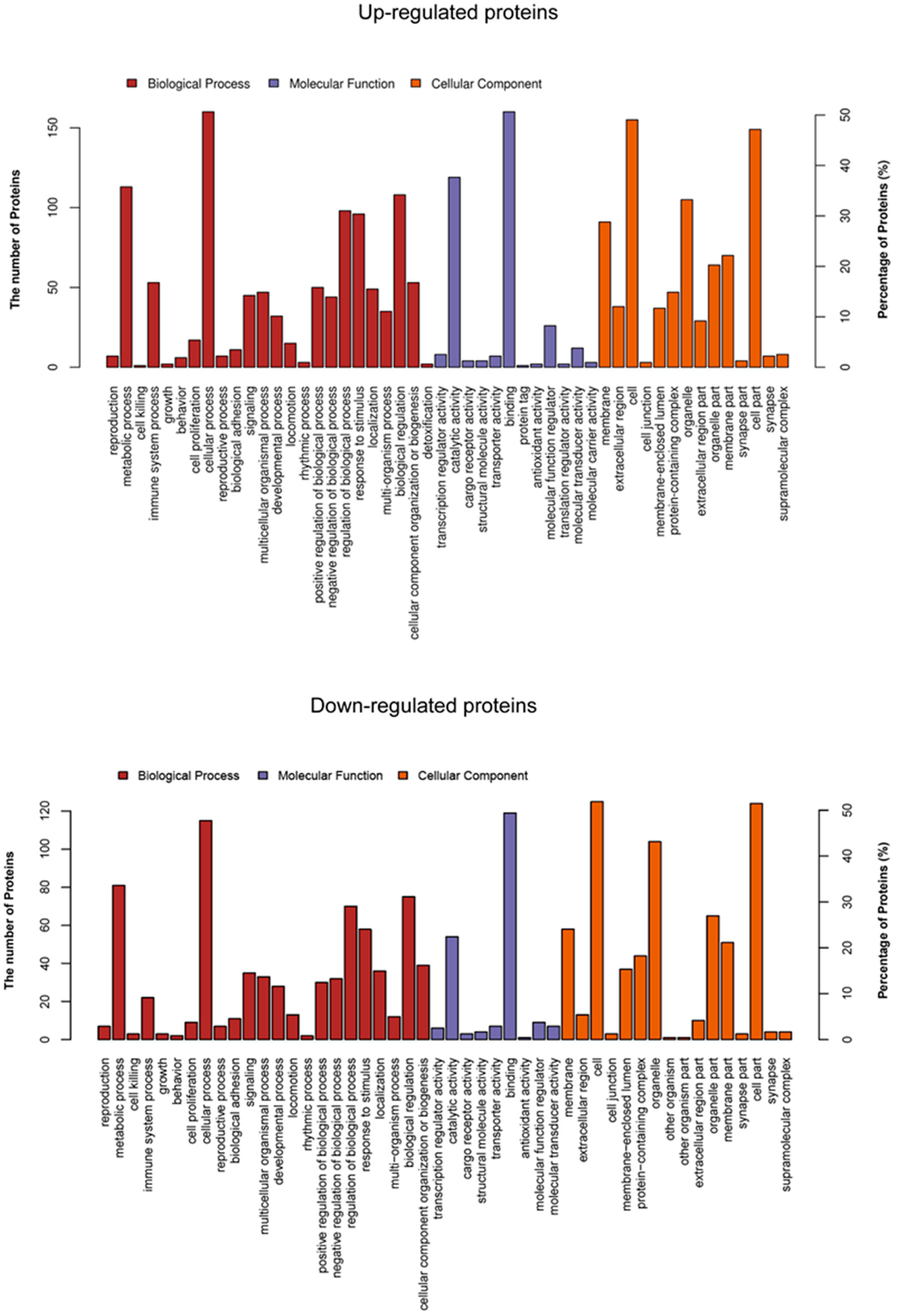
3.4. Functional Classification of Up- and Down-Regulated Proteins in the GO Database
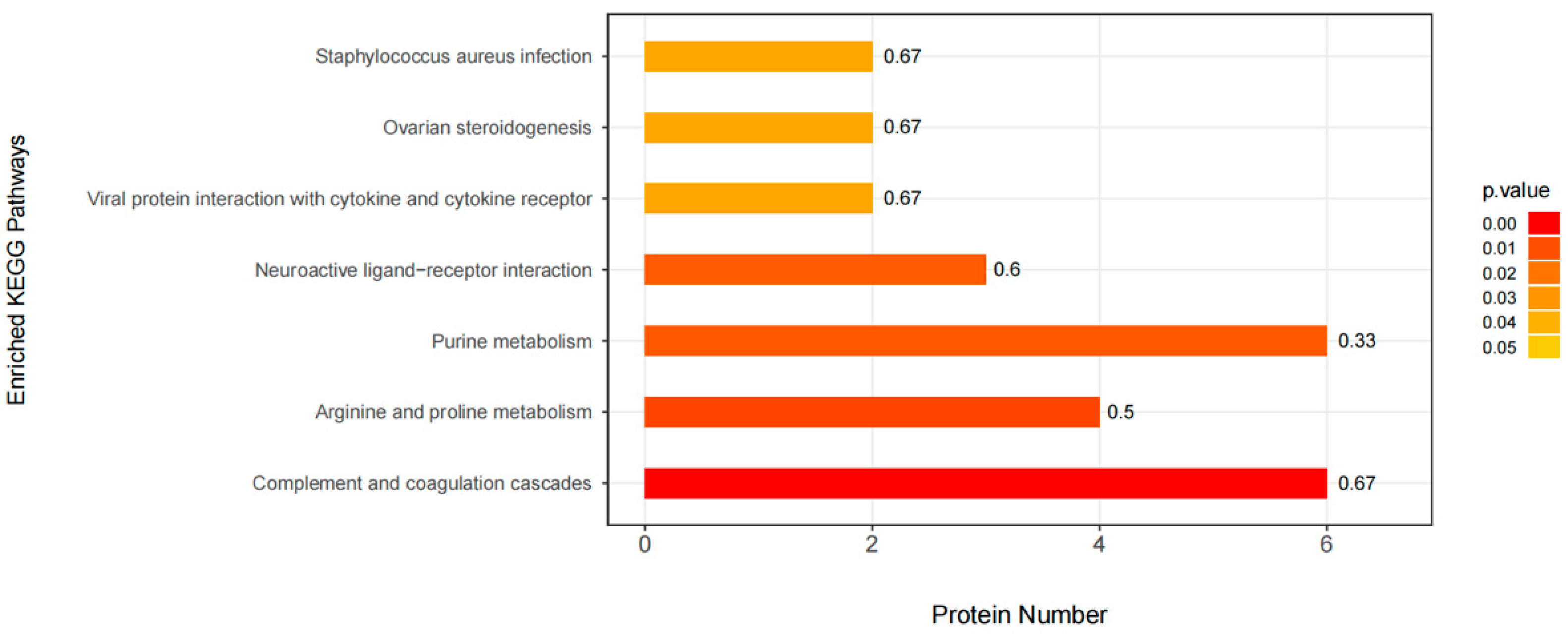
3.5. KEGG Pathway Enrichment
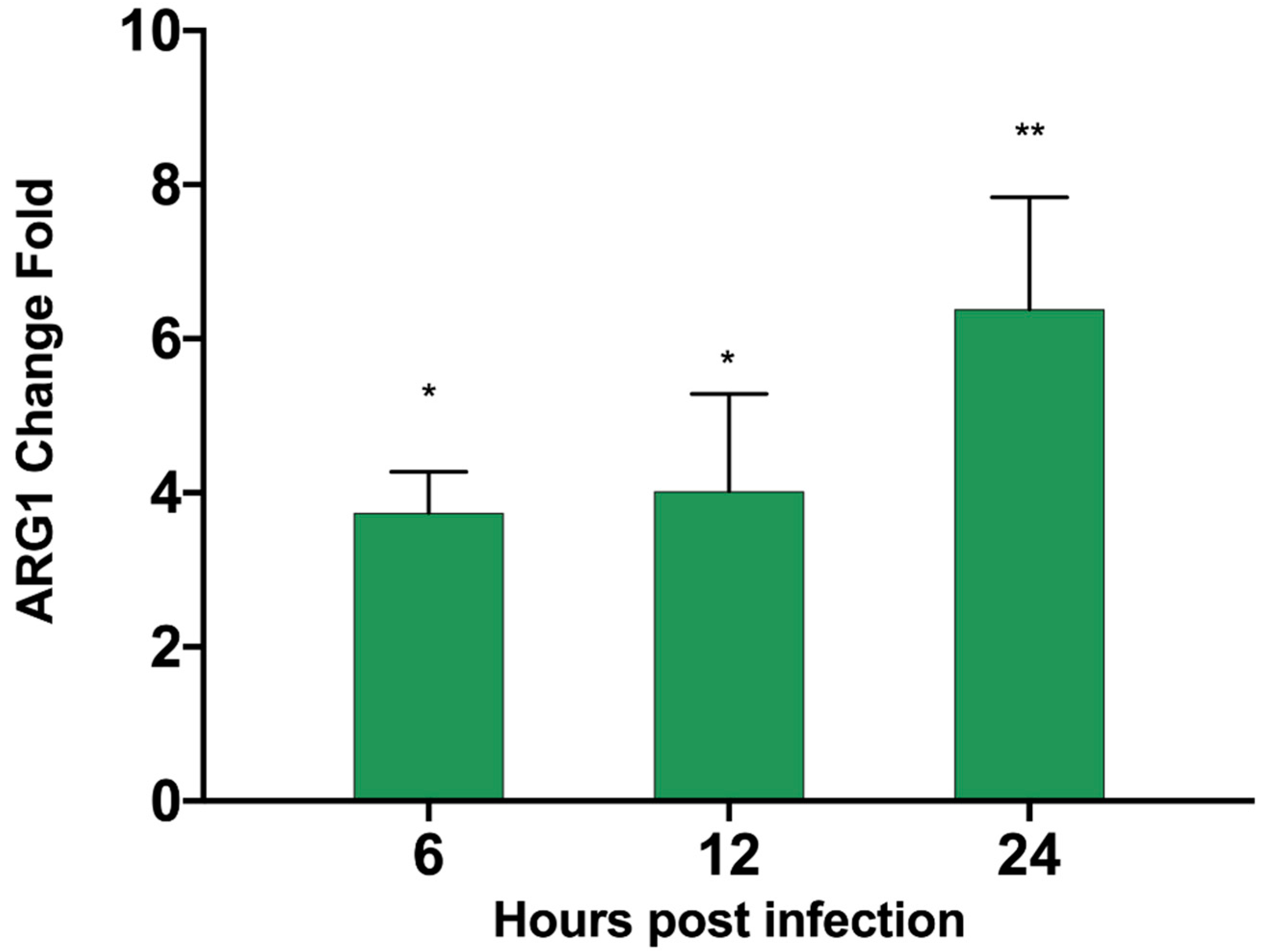
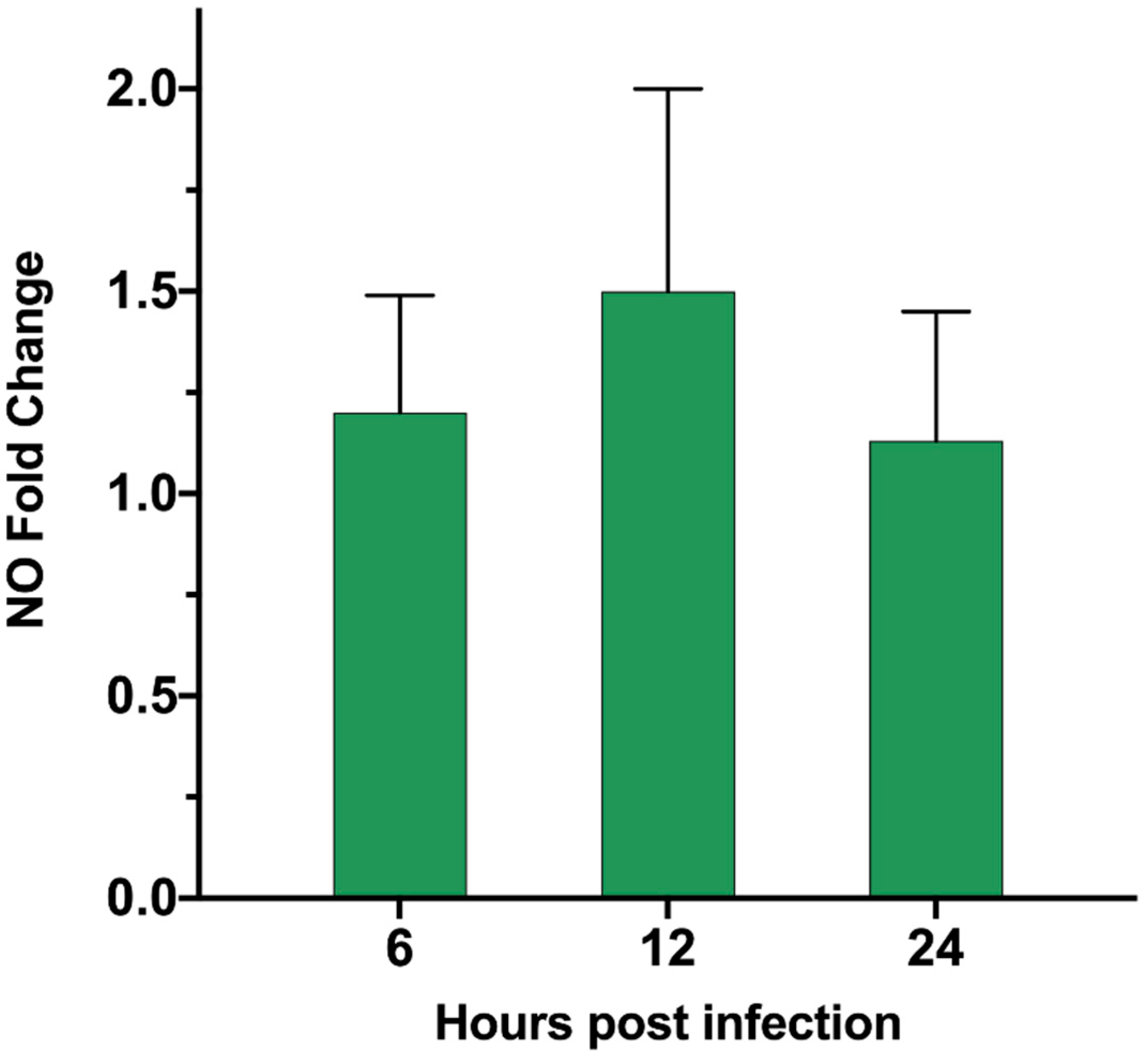
3.6. ASFV Infects PAM Cells to Promote Polyamine Upregulation Through ARG1
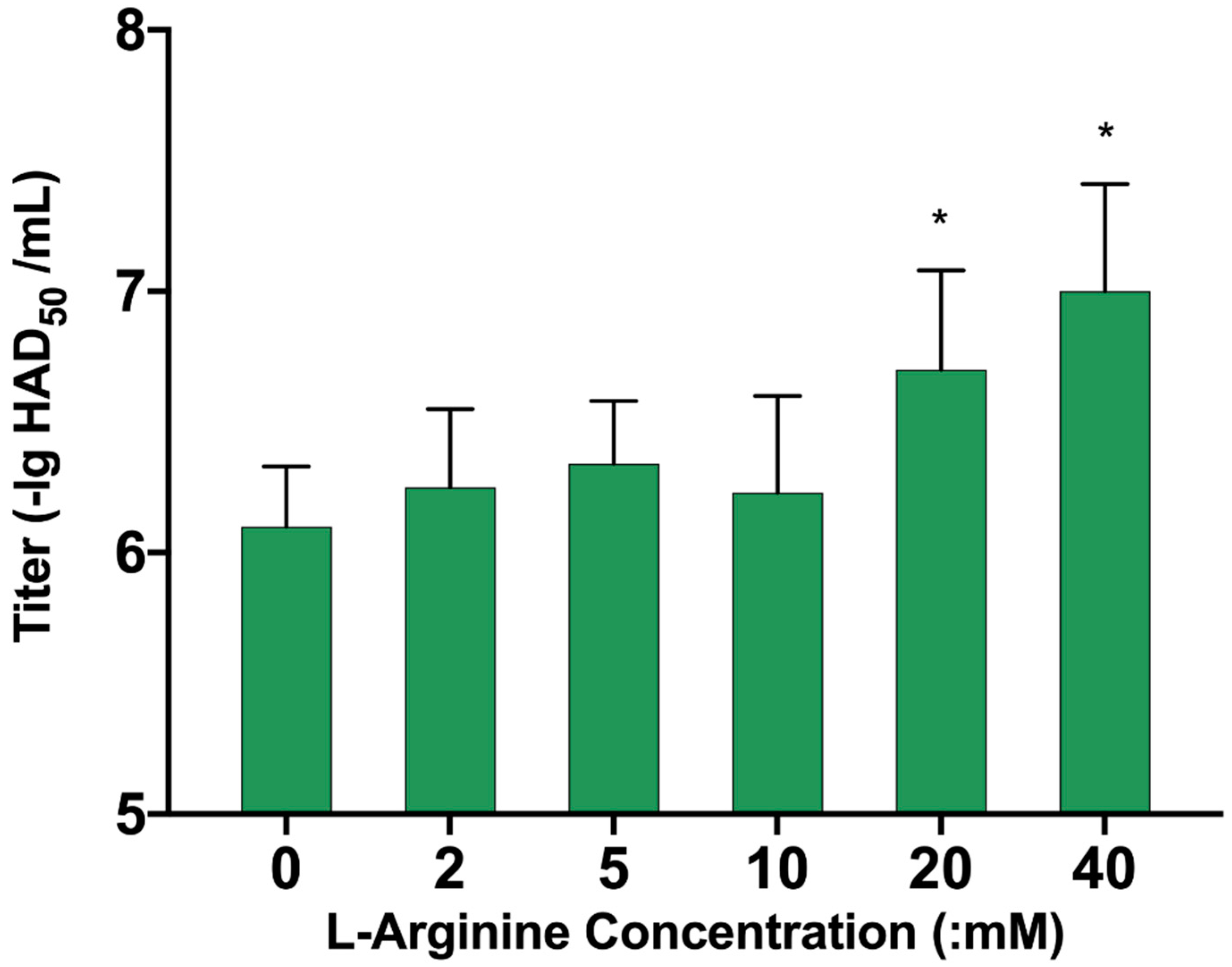
3.7. L-Arginine Can Promote ASFV Replication
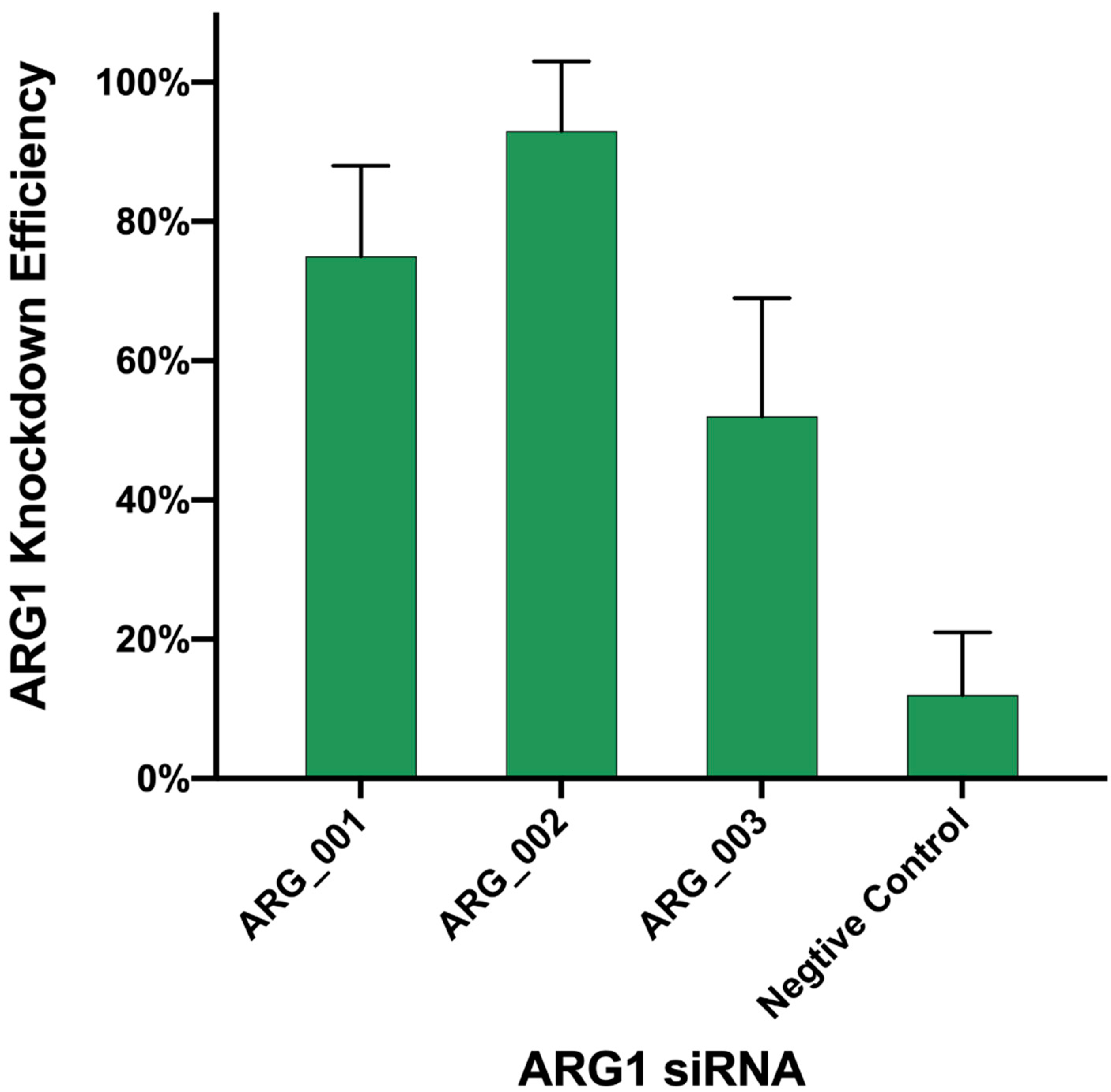
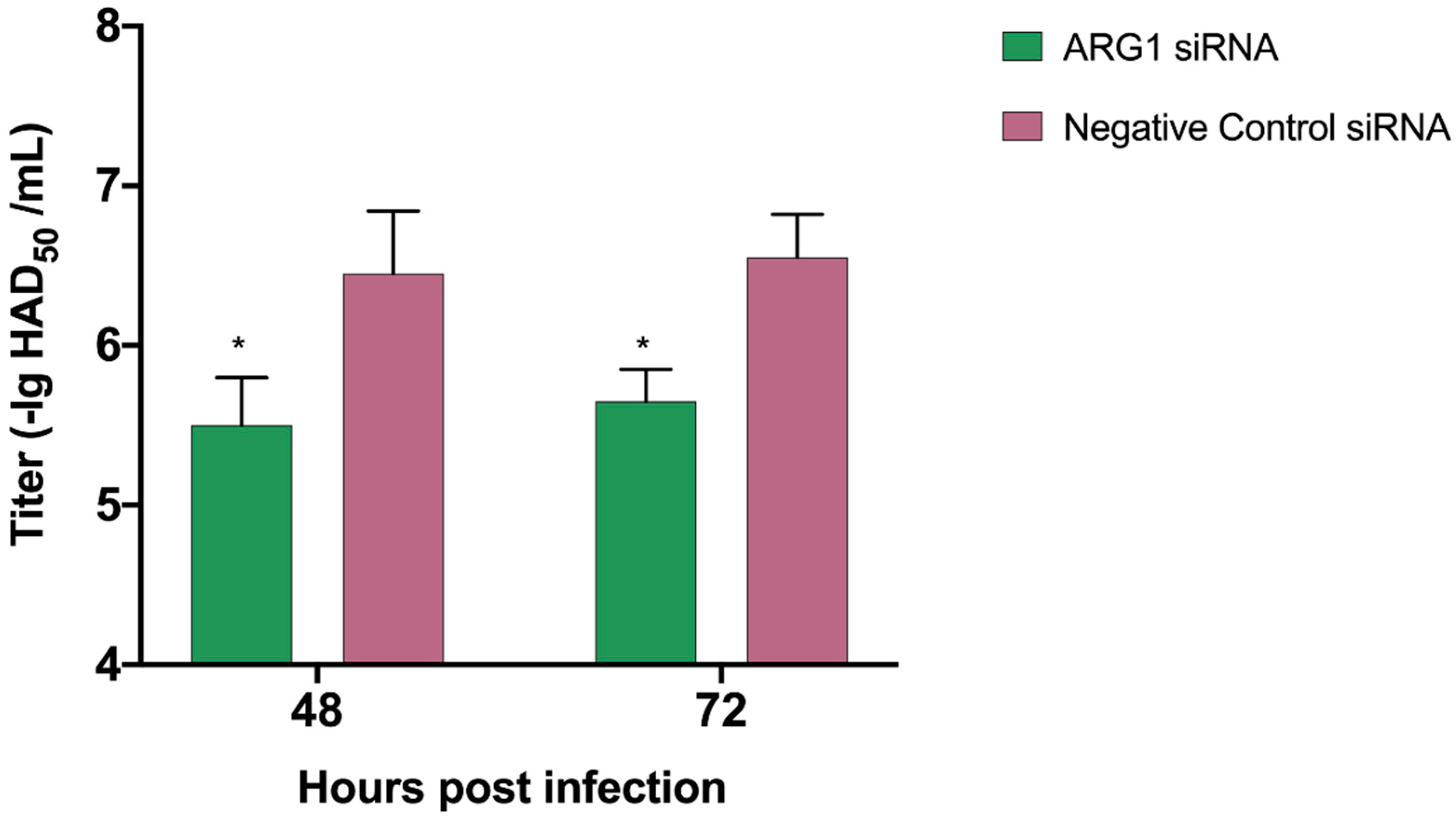
3.8. Knock Down of ARG1 Gene Affects ASFV Replication
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Martins, C.; Boinas, F.; Iacolina, L.; Ruiz-Fons, F.; Gavier-Widén, D. 1. African swine fever (ASF), the pig health challenge of the century. In Understanding and Combatting African Swine Fever; Wageningen Academic Publishers: Wageningen, The Netherlands, 2021; pp. 11–24. [Google Scholar]
- Ge, S.; Li, J.; Fan, X.; Liu, F.; Li, L.; Wang, Q.; Ren, W.; Bao, J.; Liu, C.; Wang, H.; et al. Molecular Characterization of African Swine Fever Virus, China, 2018. Emerg. Infect. Dis. 2018, 24, 2131–2133. [Google Scholar] [CrossRef] [Green Version]
- Chapman, D.A.G.; Tcherepanov, V.; Upton, C.; Dixon, L.K. Comparison of the genome sequences of non-pathogenic and pathogenic African swine fever virus isolates. J. Gen. Virol. 2008, 89, 397–408. [Google Scholar] [CrossRef] [PubMed]
- de Villiers, E.P.; Gallardo, C.; Arias, M.; da Silva, M.; Upton, C.; Martin, R.; Bishop, R.P. Phylogenomic analysis of 11 complete African swine fever virus genome sequences. Virology 2010, 400, 128–136. [Google Scholar] [CrossRef] [Green Version]
- Gallagher, J.R.; Harris, A.K. Cryo-EM cools down swine fever. J. Biol. Chem. 2020, 295, 13–14. [Google Scholar] [CrossRef] [Green Version]
- Montoya, M.; Franzoni, G.; Pérez-Nuñez, D.; Revilla, Y.; Galindo, I.; Alonso, C.; Netherton, C.; Blohm, U. 3. Immune responses against African swine fever virus infection. In Understanding and Combatting African Swine Fever; Wageningen Academic Publishers: Wageningen, The Netherlands, 2021; pp. 63–85. [Google Scholar]
- Wang, X.; Wu, J.; Wu, Y.; Chen, H. Inhibition of cGAS-STING-TBK1 signaling pathway by DP96R of ASFV China 2018/1. Biochem. Biophys. Res. Commun. 2018, 506, 437–443. [Google Scholar] [CrossRef]
- Correia, S.; Ventura, S.; Parkhouse, R.M. Identification and utility of innate immune system evasion mechanisms of ASFV. Virus Res. 2013, 173, 87–100. [Google Scholar] [CrossRef]
- Brun, A.; Rivas, C.; Esteban, M.; Escribano, J.M.; Alonso, C. African Swine Fever Virus GeneA179L, a Viral Homologue ofbcl-2, Protects Cells from Programmed Cell Death. Virology 1996, 225, 227–230. [Google Scholar] [CrossRef] [Green Version]
- Revilla, Y.; Cebrián, A.; Baixeras, E.; Martínez-A, C.; Viñuela, E.; Salas, M.L. Inhibition of Apoptosis by the African Swine Fever Virus Bcl-2 Homologue: Role of the BH1 Domain. Virology 1997, 228, 400–404. [Google Scholar] [CrossRef] [Green Version]
- Nogal, M.L.; de Buitrago, G.G.; Rodríguez, C.; Cubelos, B.; Carrascosa, A.L.; Salas, M.L.; Revilla, Y. African Swine Fever Virus IAP Homologue Inhibits Caspase Activation and Promotes Cell Survival in Mammalian Cells. J. Virol. 2001, 75, 2535–2543. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez, C.I.; Nogal, M.L.; Carrascosa, A.L.; Salas, M.L.; Fresno, M.; Revilla, Y. African Swine Fever Virus IAP-Like Protein Induces the Activation of Nuclear Factor Kappa B. J. Virol. 2002, 76, 3936–3942. [Google Scholar] [CrossRef] [Green Version]
- Gladue, D.P.; O’Donnell, V.; Ramirez-Medina, E.; Rai, A.; Pruitt, S.; Vuono, E.A.; Silva, E.; Velazquez-Salinas, L.; Borca, M.V. Deletion of CD2-Like (CD2v) and C-Type Lectin-Like (EP153R) Genes from African Swine Fever Virus Georgia-∆9GL Abrogates Its Effectiveness as an Experimental Vaccine. Viruses 2020, 12, 1185. [Google Scholar] [CrossRef]
- Zhang, F.; Moon, A.; Childs, K.; Goodbourn, S.; Dixon, L.K. The African Swine Fever Virus DP71L Protein Recruits the Protein Phosphatase 1 Catalytic Subunit to Dephosphorylate eIF2α and Inhibits CHOP Induction but Is Dispensable for These Activities during Virus Infection. J. Virol. 2010, 84, 10681–10689. [Google Scholar] [CrossRef] [Green Version]
- Silk, R.N.; Bowick, G.C.; Abrams, C.C.; Dixon, L.K. African swine fever virus A238L inhibitor of NF-kappaB and of calcineurin phosphatase is imported actively into the nucleus and exported by a CRM1-mediated pathway. J. Gen. Virol. 2007, 88, 411–419. [Google Scholar] [CrossRef]
- Borca, M.V.; O’Donnell, V.; Holinka, L.G.; Ramírez-Medina, E.; Clark, B.A.; Vuono, E.A.; Berggren, K.; Alfano, M.; Carey, L.B.; Richt, J.A.; et al. The L83L ORF of African swine fever virus strain Georgia encodes for a non-essential gene that interacts with the host protein IL-1beta. Virus Res. 2018, 249, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, L.; De Lara, F.C.-M.; Mulas, J.M.D.L.; Gómez-Villamandos, J.C.; Sierra, M.A.; Villeda, C.J.; Wilkinson, P.J. Ultrastructural changes related to the lymph node haemorrhages in acute African swine fever. Res. Veter Sci. 1997, 62, 199–204. [Google Scholar] [CrossRef]
- Sierra, M.; Carrasco, L.; Gómez-Villamandos, J.; Mulas, J.M.D.L.; Mendez, A.; Jover, A. Pulmonary intravascular macrophages in lungs of pigs inoculated with african swine fever virus of differing virulence. J. Comp. Pathol. 1990, 102, 323–334. [Google Scholar] [CrossRef]
- Rodriguez, J.M.; Salas, M.L.; Santaren, J.F. African swine fever virus-induced polypeptides in porcine alveolar macrophages and in Vero cells: Two-dimensional gel analysis. Proteomics 2001, 1, 1447–1456. [Google Scholar] [CrossRef]
- Alfonso, P.; Rivera, J.; Hernáez, B.; Alonso, C.; Escribano, J.M. Identification of cellular proteins modified in response to African swine fever virus infection by proteomics. Proteomics 2004, 4, 2037–2046. [Google Scholar] [CrossRef] [PubMed]
- Keßler, C.; Forth, J.H.; Keil, G.M.; Mettenleiter, T.C.; Blome, S.; Karger, A. The intracellular proteome of African swine fever virus. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Herrera-Uribe, J.; Jiménez-Marín, Á.; Lacasta, A.; Monteagudo, P.L.; Pina-Pedrero, S.; Rodríguez, F.; Moreno, Á.; Garrido, J.J. Comparative proteomic analysis reveals different responses in porcine lymph nodes to virulent and attenuated homologous African swine fever virus strains. Veter Res. 2018, 49, 90. [Google Scholar] [CrossRef] [Green Version]
- McAlister, G.C.; Huttlin, E.L.; Haas, W.; Ting, L.; Jedrychowski, M.P.; Rogers, J.C.; Kuhn, K.; Pike, I.; Grothe, R.A.; Blethrow, J.D.; et al. Increasing the multiplexing capacity of TMTs using reporter ion isotopologues with isobaric masses. Anal. Chem. 2012, 84, 7469–7478. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Yu, Y.; Montani, J.P.; Ming, X.F.; Yang, Z. Arginase-I enhances vascular endothelial inflammation and senescence through eNOS-uncoupling. BMC Res. Notes 2017, 10, 82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michael, A.J. Polyamines in Eukaryotes, Bacteria, and Archaea. J. Biol. Chem. 2016, 291, 14896–14903. [Google Scholar] [CrossRef] [Green Version]
- Mastrodomenico, V.; Esin, J.J.; Graham, M.L.; Tate, P.M.; Hawkins, G.M.; Sandler, Z.J.; Rademacher, D.J.; Kicmal, T.M.; Dial, C.; Mounce, B.C. Polyamine Depletion Inhibits Bunyavirus Infection via Generation of Noninfectious Interfering Virions. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [Green Version]
- Li, X.-K.; Lu, Q.-B.; Chen, W.-W.; Xu, W.; Liu, R.; Zhang, S.-F.; Du, J.; Li, H.; Yao, K.; Zhai, D.; et al. Arginine deficiency is involved in thrombocytopenia and immunosuppression in severe fever with thrombocytopenia syndrome. Sci. Transl. Med. 2018, 10, eaat4162. [Google Scholar] [CrossRef] [Green Version]
- Madhubala, R. Thin-Layer Chromatographic Method for Assaying Polyamines. Polyam. Protoc. 1998, 79, 131–136. [Google Scholar] [CrossRef]
- Liu, Z.G.; Hsu, H.; Goeddel, D.V. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 1996, 87, 565–576. [Google Scholar] [CrossRef] [Green Version]
- Fotin-Mleczek, M.; Henkler, F.; Hausser, A.; Glauner, H.; Samel, D.; Graness, A.; Scheurich, P.; Mauri, D.; Wajant, H. Tumor necrosis factor receptor-associated factor (TRAF) 1 regulates CD40-induced TRAF2-mediated NF-kappaB activation. J. Biol. Chem. 2004, 279, 677–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hacker, H.; Redecke, V.; Blagoev, B.; Kratchmarova, I.; Hsu, L.-C.; Wang, G.G.; Kamps, M.P.; Raz, E.; Wagner, H.; Häcker, G.; et al. Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 2006, 439, 204–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oganesyan, G.; Saha, S.K.; Guo, B.; He, J.Q.; Shahangian, A.; Zarnegar, B.; Perry, A.; Cheng, G. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nat. Cell Biol. 2005, 439, 208–211. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Cheng, G.; Baltimore, D. Targeted Disruption of TRAF3 Leads to Postnatal Lethality and Defective T-Dependent Immune Responses. Immunity 1996, 5, 407–415. [Google Scholar] [CrossRef] [Green Version]
- Shih, C.-H.; Van Eeden, S.F.; Goto, Y.; Hogg, J.C. CCL23/myeloid progenitor inhibitory factor-1 inhibits production and release of polymorphonuclear leukocytes and monocytes from the bone marrow. Exp. Hematol. 2005, 33, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Arruda-Silva, F.; Bianchetto-Aguilera, F.; Gasperini, S.; Polletti, S.; Cosentino, E.; Tamassia, N.; Cassatella, M.A. Human Neutrophils Produce CCL23 in Response to Various TLR-Agonists and TNFalpha. Front Cell Infect Microbiol. 2017, 7, 176. [Google Scholar] [CrossRef] [Green Version]
- Touchman, J.W.; Anikster, Y.; Dietrich, N.L.; Maduro, V.V.B.; McDowell, G.; Shotelersuk, V.; Bouffard, G.G.; Beckstrom-Sternberg, S.M.; Gahl, W.A.; Green, E.D. The Genomic Region Encompassing the Nephropathic Cystinosis Gene (CTNS): Complete Sequencing of a 200-kb Segment and Discovery of a Novel Gene within the Common Cystinosis-Causing Deletion. Genome Res. 2000, 10, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Kardon, T.; Stroobant, V.; Veiga-Da-Cunha, M.; Van Schaftingen, E. Characterization of mammalian sedoheptulokinase and mechanism of formation of erythritol in sedoheptulokinase deficiency. FEBS Lett. 2008, 582, 3330–3334. [Google Scholar] [CrossRef] [Green Version]
- Wamelink, M.M.; Struys, E.A.; Jansen, E.E.; Levtchenko, E.N.; Zijlstra, F.S.; Engelke, U.; Blom, H.J.; Jakobs, C.; Wevers, R.A. Sedoheptulokinase deficiency due to a 57-kb deletion in cystinosis patients causes urinary accumulation of sedoheptulose: Elucidation of the CARKL gene. Hum. Mutat. 2008, 29, 532–536. [Google Scholar] [CrossRef]
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C.; et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012, 15, 813–826. [Google Scholar] [CrossRef] [Green Version]
- Latchman, Y.E.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.J.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat. Immunol. 2001, 2, 261–268. [Google Scholar] [CrossRef]
- Huang, Z.; Su, G.-F.; Hu, W.-J.; Bi, X.-X.; Zhang, L.; Wan, G. The study on expression of CIAPIN1 interfering hepatocellular carcinoma cell proliferation and its mechanisms. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 3054–3060. [Google Scholar]
- Shibayama, H.; Takai, E.; Matsumura, I.; Kouno, M.; Morii, E.; Kitamura, Y.; Takeda, J.; Kanakura, Y. Identification of a Cytokine-induced Antiapoptotic Molecule Anamorsin Essential for Definitive Hematopoiesis. J. Exp. Med. 2004, 199, 581–592. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Zhao, W.; Zheng, Q.; Chen, T.; Qi, J.; Li, G.; Tong, T. Ribosomal L1 domain and lysine-rich region are essential for CSIG/RSL1D1 to regulate proliferation and senescence. Biochem. Biophys. Res. Commun. 2016, 469, 593–598. [Google Scholar] [CrossRef]
- Ma, L.; Chang, N.; Guo, S.; Li, Q.; Zhang, Z.; Wang, W.; Tong, T. CSIG Inhibits PTEN Translation in Replicative Senescence. Mol. Cell. Biol. 2008, 28, 6290–6301. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Q.; Yuan, F.; Lu, F.; Zhang, B.; Chen, T.; Chen, X.; Cheng, Y.; Li, N.; Ma, L.; Tong, T. CSIG promotes hepatocellular carcinoma proliferation by activating c-MYC expression. Oncotarget 2015, 6, 4733–4744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, N.; Zhao, G.; Chen, T.; Xue, L.; Ma, L.; Niu, J.; Tong, T. Nucleolar protein CSIG is required for p33ING1 function in UV-induced apoptosis. Cell Death Dis. 2012, 3, e283. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.-T.; Ma, L.-W.; Tong, T.-J. Advance research on cellular senescence-inhibited gene (CSIG). Sheng Li Ke Xue Jin Zhan 2012, 43, 291–293. [Google Scholar]
- Xie, N.; Ma, L.; Zhu, F.; Zhao, W.; Tian, F.; Yuan, F.; Fu, J.; Huang, D.; Lv, C.; Tong, T. Regulation of the MDM2-p53 pathway by the nucleolar protein CSIG in response to nucleolar stress. Sci. Rep. 2016, 6, 36171. [Google Scholar] [CrossRef] [Green Version]
- Hamacher-Brady, A.; Brady, N.R. Mitophagy programs: Mechanisms and physiological implications of mitochondrial targeting by autophagy. Cell. Mol. Life Sci. 2016, 73, 775–795. [Google Scholar] [CrossRef] [Green Version]
- Tracy, K.; Dibling, B.C.; Spike, B.T.; Knabb, J.R.; Schumacker, P.; Macleod, K.F. BNIP3 Is an RB/E2F Target Gene Required for Hypoxia-Induced Autophagy. Mol. Cell. Biol. 2007, 27, 6229–6242. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.-Y.; Zhu, S.-H.; Li, V.; Gibson, S.; Xu, X.; Kong, J.-M. BNIP3 Interacting with LC3 Triggers Excessive Mitophagy in Delayed Neuronal Death in Stroke. CNS Neurosci. Ther. 2014, 20, 1045–1055. [Google Scholar] [CrossRef]
- Plenchette, S.; Cheung, H.H.; Fong, W.G.; LaCasse, E.C.; Korneluk, R.G. The role of XAF1 in cancer. Curr. Opin. Investig. Drugs 2007, 8, 469–476. [Google Scholar]
- Laird, M.E.; Mohsen, A.; Duffy, D.; Mamdouh, R.; LeFouler, L.; Casrouge, A.; El-Daly, M.; Rafik, M.; Abdel-Hamid, M.; Soulier, A.; et al. Apolipoprotein H expression is associated with IL28B genotype and viral clearance in hepatitis C virus infection. J. Hepatol. 2014, 61, 770–776. [Google Scholar] [CrossRef]
- Henderson, J.P.; Byun, J.; Takeshita, J.; Heinecke, J.W. Phagocytes Produce 5-Chlorouracil and 5-Bromouracil, Two Mutagenic Products of Myeloperoxidase, in Human Inflammatory Tissue. J. Biol. Chem. 2003, 278, 23522–23528. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Q.; Blount, B.C.; Ames, B.N. 5-Chlorouracil, a Marker of DNA Damage from Hypochlorous Acid during Inflammation. J. Biol. Chem. 2003, 278, 32834–32840. [Google Scholar] [CrossRef] [Green Version]
- Takeshita, J.; Byun, J.; Nhan, T.Q.; Pritchard, D.K.; Pennathur, S.; Schwartz, S.M.; Chait, A.; Heinecke, J.W. Myeloperoxidase generates 5-chlorouracil in human atherosclerotic tissue: A potential pathway for somatic mutagenesis by macrophages. J. Biol. Chem. 2006, 281, 3096–3104. [Google Scholar] [CrossRef] [Green Version]
- Núñez-Navarro, N.E.; Santana, F.M.; Parra, L.P.; Zacconi, F.C. Surfing the Blood Coagulation Cascade: Insight into the Vital Factor Xa. Curr. Med. Chem. 2019, 26, 3175–3200. [Google Scholar] [CrossRef]
- Liu, C.; Mao, L.; Ping, Z.; Jiang, T.; Wang, C.; Chen, Z.; Li, Z.; Li, J. Serum Protein KNG1, APOC3, and PON1 as Potential Biomarkers for Yin-Deficiency-Heat Syndrome. Evid. Based Complement. Altern. Med. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Brunel, H.; Massanet, R.; Martinez-Perez, A.; Ziyatdinov, A.; Fernández, J.M.S.; Souto, J.C.; Perera, A.; Soria, J.M. The Central Role of KNG1 Gene as a Genetic Determinant of Coagulation Pathway-Related Traits: Exploring Metaphenotypes. PLoS ONE 2016, 11, e0167187. [Google Scholar] [CrossRef]
- Yue, Y.; Liu, S.; Han, X.; Xiao, L.; Huang, Q.; Li, S.; Zhuang, K.; Yang, M.; Zou, C.; Fu, Y. Pathogenic variants of PROC gene caused type I activity deficiency in a familial Chinese venous thrombosis. J. Cell. Mol. Med. 2019, 23, 7099–7104. [Google Scholar] [CrossRef] [Green Version]
- Wade, K.C.; Guttentag, S.H.; Gonzales, L.W.; Maschhoff, K.L.; Gonzales, J.; Kolla, V.; Singhal, S.; Ballard, P.L. Gene Induction during Differentiation of Human Pulmonary Type II Cells In Vitro. Am. J. Respir. Cell Mol. Biol. 2006, 34, 727–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brasch, F.; Johnen, G.; Winn-Brasch, A.; Guttentag, S.H.; Schmiedl, A.; Kapp, N.; Suzuki, Y.; Müller, K.M.; Richter, J.; Hawgood, S.; et al. Surfactant Protein B in Type II Pneumocytes and Intra-Alveolar Surfactant Forms of Human Lungs. Am. J. Respir. Cell Mol. Biol. 2004, 30, 449–458. [Google Scholar] [CrossRef]
- Hobi, N.; Giolai, M.; Olmeda, B.; Miklavc, P.; Felder, E.; Walther, P.; Dietl, P.; Frick, M.; Pérez-Gil, J.; Haller, T. A small key unlocks a heavy door: The essential function of the small hydrophobic proteins SP-B and SP-C to trigger adsorption of pulmonary surfactant lamellar bodies. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2016, 1863, 2124–2134. [Google Scholar] [CrossRef] [PubMed]
- Albert, R.K. The Role of Ventilation-induced Surfactant Dysfunction and Atelectasis in Causing Acute Respiratory Distress Syndrome. Am. J. Respir. Crit. Care Med. 2012, 185, 702–708. [Google Scholar] [CrossRef]
- Sánchez-Cordón, P.; Vidaña, B.; Neimanis, A.; Núñez, A.; Wikström, E.; Gavier-Widén, D. 4. Pathology of African swine fever. In Understanding and Combatting African Swine Fever; Wageningen Academic Publishers: Wageningen, The Netherlands, 2021; pp. 87–139. [Google Scholar]
- Burrack, K.S.; Morrison, T.E. The Role of Myeloid Cell Activation and Arginine Metabolism in the Pathogenesis of Virus-Induced Diseases. Front. Immunol. 2014, 5, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sautter, C.A.; Auray, G.; Python, S.; Liniger, M.; Summerfield, A. Phenotypic and functional modulations of porcine macrophages by interferons and interleukin-4. Dev. Comp. Immunol. 2018, 84, 181–192. [Google Scholar] [CrossRef]
- Franzoni, G.; Razzuoli, E.; Giudici, S.D.; Carta, T.; Galleri, G.; Zinellu, S.; Ledda, M.; Angioi, P.; Modesto, P.; Graham, S.P.; et al. Comparison of Macrophage Responses to African Swine Fever Viruses Reveals that the NH/P68 Strain is Associated with Enhanced Sensitivity to Type I IFN and Cytokine Responses from Classically Activated Macrophages. Pathogens 2020, 9, 209. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| ARG1-qF | TGTCTTCCGTTCAGTAGGTGG |
| ARG1-qR | TACACCAGAGTCCTCCAGCC |
| Target Gene | Target Sequence |
|---|---|
| ARG1_001 | GTGGCAGAAATCAAGAAGA |
| ARG1_002 | CCTGAAACCACCTAAGTAA |
| ARG1_003 | GCTATCTACTAGGAAGAAA |
| Gene | I/M Ratio 1 | t-Test p-Value |
|---|---|---|
| Up regulated | ||
| RSAD2 | 3.220821 | 3.7108 × 10−5 |
| DDX58 | 2.410459 | 0.00020296 |
| ARG1 | 1.988772 | 0.00427627 |
| F9 | 1.797021 | 1.8209 × 10−6 |
| XAF1 | 1.752608 | 0.00781031 |
| KNG1 | 1.665106 | 0.00626593 |
| SHPK | 1.608692 | 0.01255248 |
| APOH | 1.594255 | 0.01519234 |
| CIAPIN1 | 1.569497 | 0.00012041 |
| CLU | 1.549828 | 0.00127142 |
| TRAF3 | 1.543032 | 0.00051584 |
| PD-L2 | 1.402388 | 0.00099183 |
| C5AR1 | 1.343329 | 0.00063514 |
| PROC | 1.320784 | 0.00276507 |
| CAMK2D | 1.288763 | 0.00040577 |
| TNFAIP6 | 1.210334 | 0.0297009 |
| Down regulated | ||
| SFTPB | 0.60814 | 0.01322343 |
| CCL23 | 0.623738 | 0.0098507 |
| BNIP3 | 0.74174 | 0.00138866 |
| RSL1D1 | 0.819272 | 0.01253252 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ai, Q.; Lin, X.; Xie, H.; Li, B.; Liao, M.; Fan, H. Proteome Analysis in PAM Cells Reveals That African Swine Fever Virus Can Regulate the Level of Intracellular Polyamines to Facilitate Its Own Replication through ARG1. Viruses 2021, 13, 1236. https://doi.org/10.3390/v13071236
Ai Q, Lin X, Xie H, Li B, Liao M, Fan H. Proteome Analysis in PAM Cells Reveals That African Swine Fever Virus Can Regulate the Level of Intracellular Polyamines to Facilitate Its Own Replication through ARG1. Viruses. 2021; 13(7):1236. https://doi.org/10.3390/v13071236
Chicago/Turabian StyleAi, Qiangyun, Xiwei Lin, Hangao Xie, Bin Li, Ming Liao, and Huiying Fan. 2021. "Proteome Analysis in PAM Cells Reveals That African Swine Fever Virus Can Regulate the Level of Intracellular Polyamines to Facilitate Its Own Replication through ARG1" Viruses 13, no. 7: 1236. https://doi.org/10.3390/v13071236
APA StyleAi, Q., Lin, X., Xie, H., Li, B., Liao, M., & Fan, H. (2021). Proteome Analysis in PAM Cells Reveals That African Swine Fever Virus Can Regulate the Level of Intracellular Polyamines to Facilitate Its Own Replication through ARG1. Viruses, 13(7), 1236. https://doi.org/10.3390/v13071236