
DsRNA Sequencing for RNA Virus Surveillance Using Human Clinical Samples

 , , , , ,
, , , , ,  add
Show full author list
add
Show full author list
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical and Other Samples
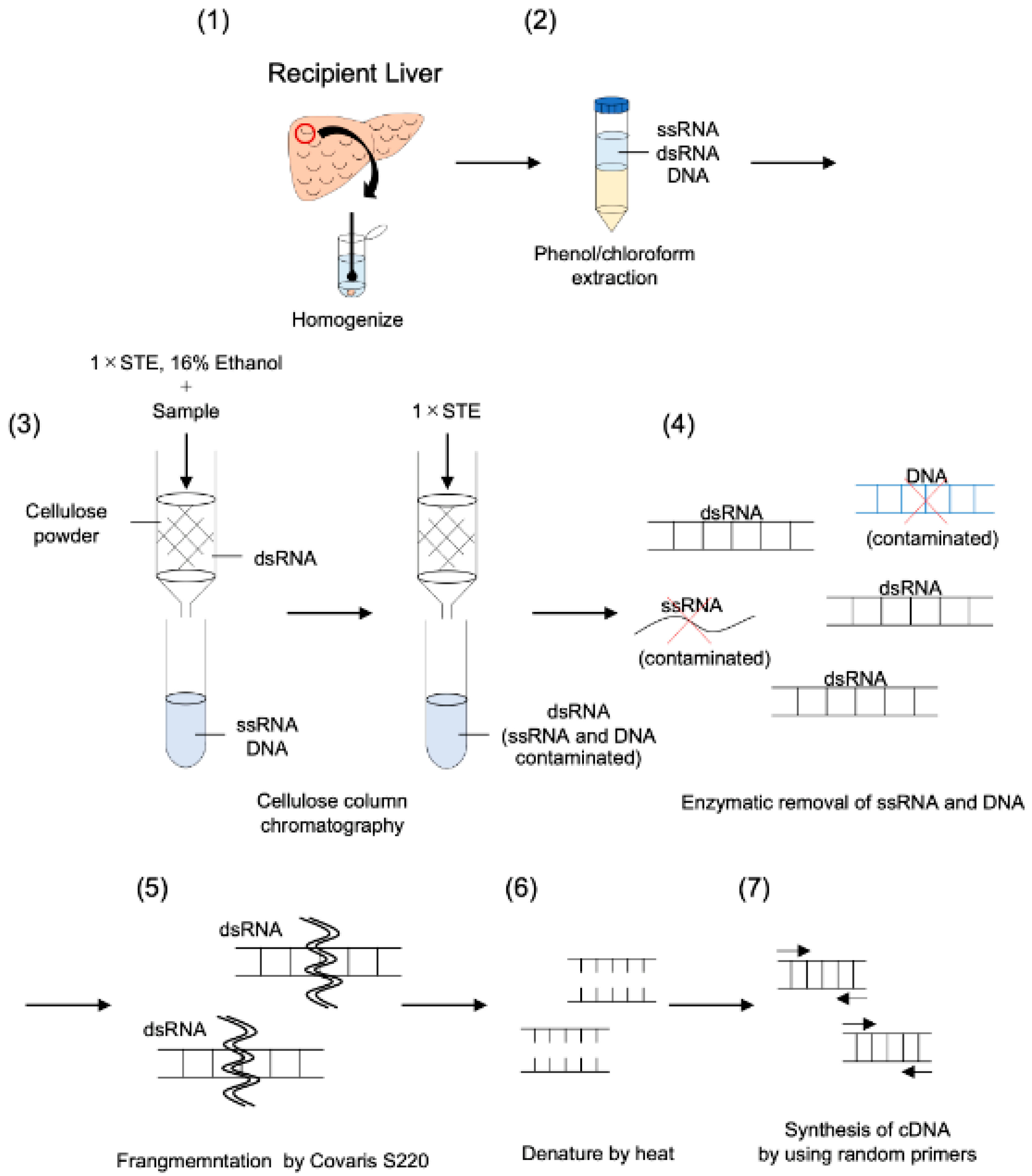
2.2. Purification of dsRNA and cDNA Synthesis Using Modified dsRNA-Seq Method
2.3. cDNA Synthesis Using the FLDS Method
2.4. Total RNA Extraction and cDNA Synthesis for Total RNA Sequencing
2.5. Illumina Sequencing, Data Assembly and Processing
2.6. Phylogenetic Tree Analysis
2.7. Virus Infection Assay
3. Results
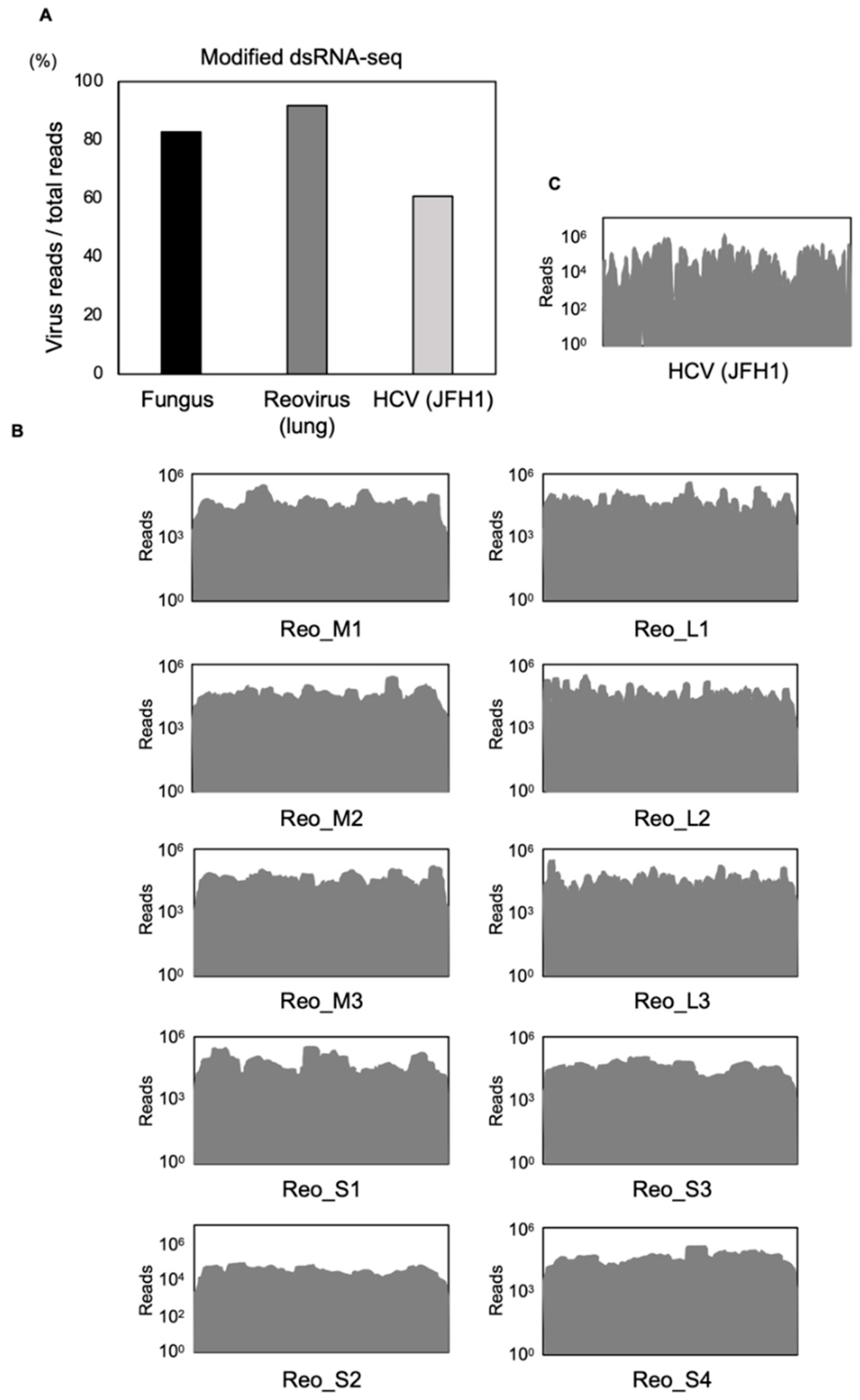
3.1. Comparison between Modified dsRNA-Seq and Other Conventional Methods
3.2. Modified dsRNA-Seq Analysis in Animal Tissues and Cultured Cells
3.3. Intracellular RNA Virus Surveillance and Library Construction
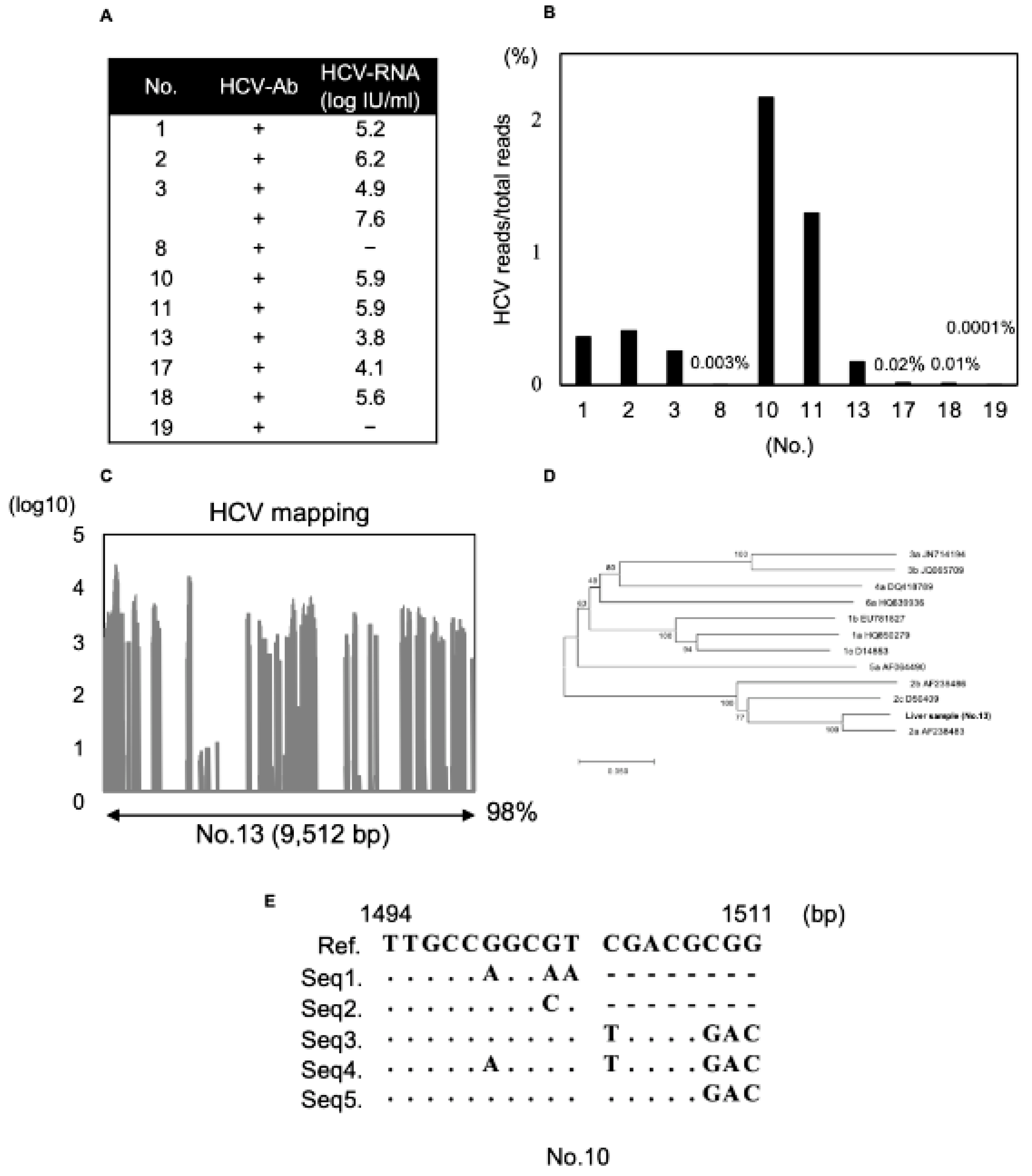
3.4. Detection of HCV Genome Using Modified dsRNA-Seq
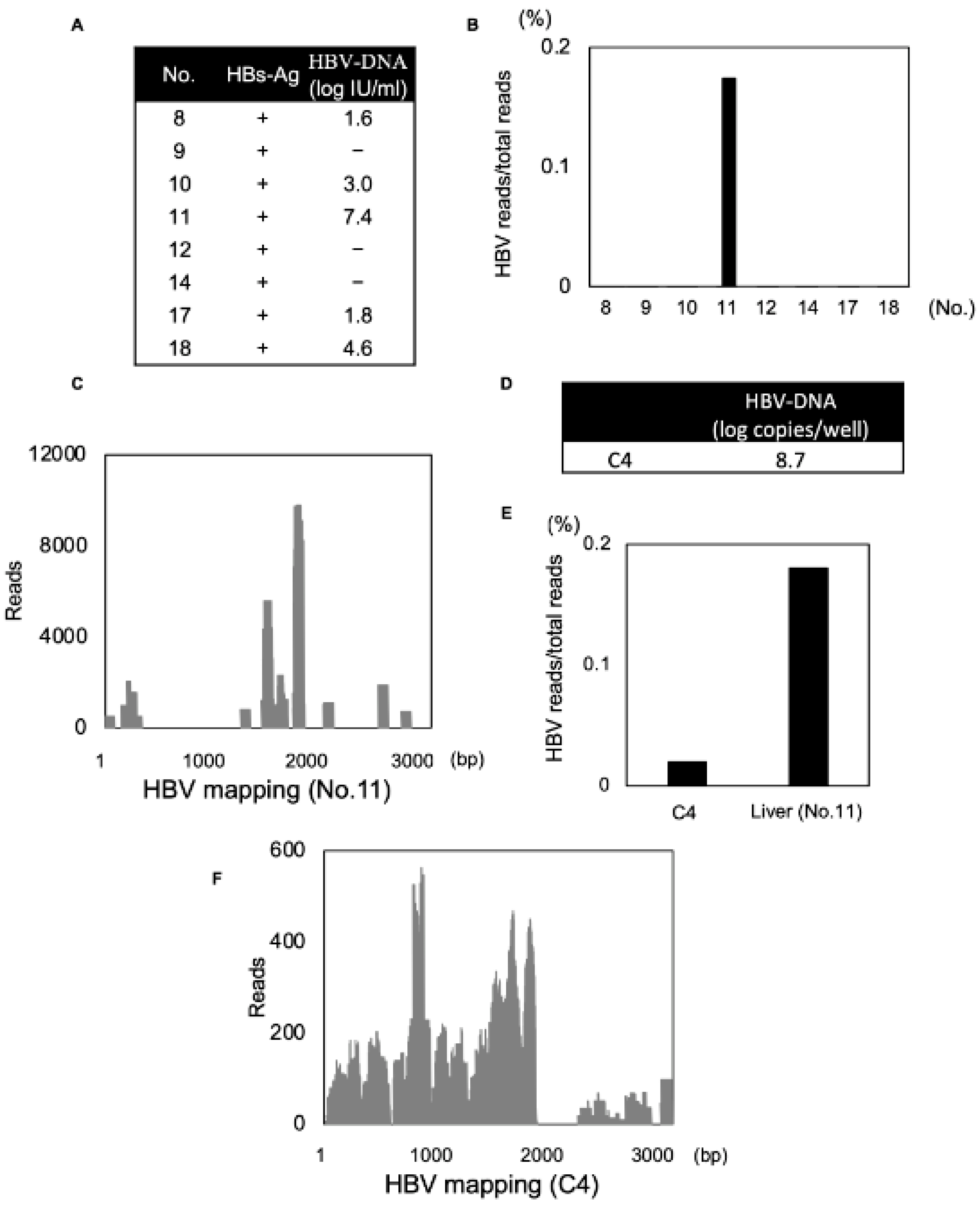
3.5. Detection of DNA Viral Genome with a Secondary Structure Using Modified dsRNA-Seq
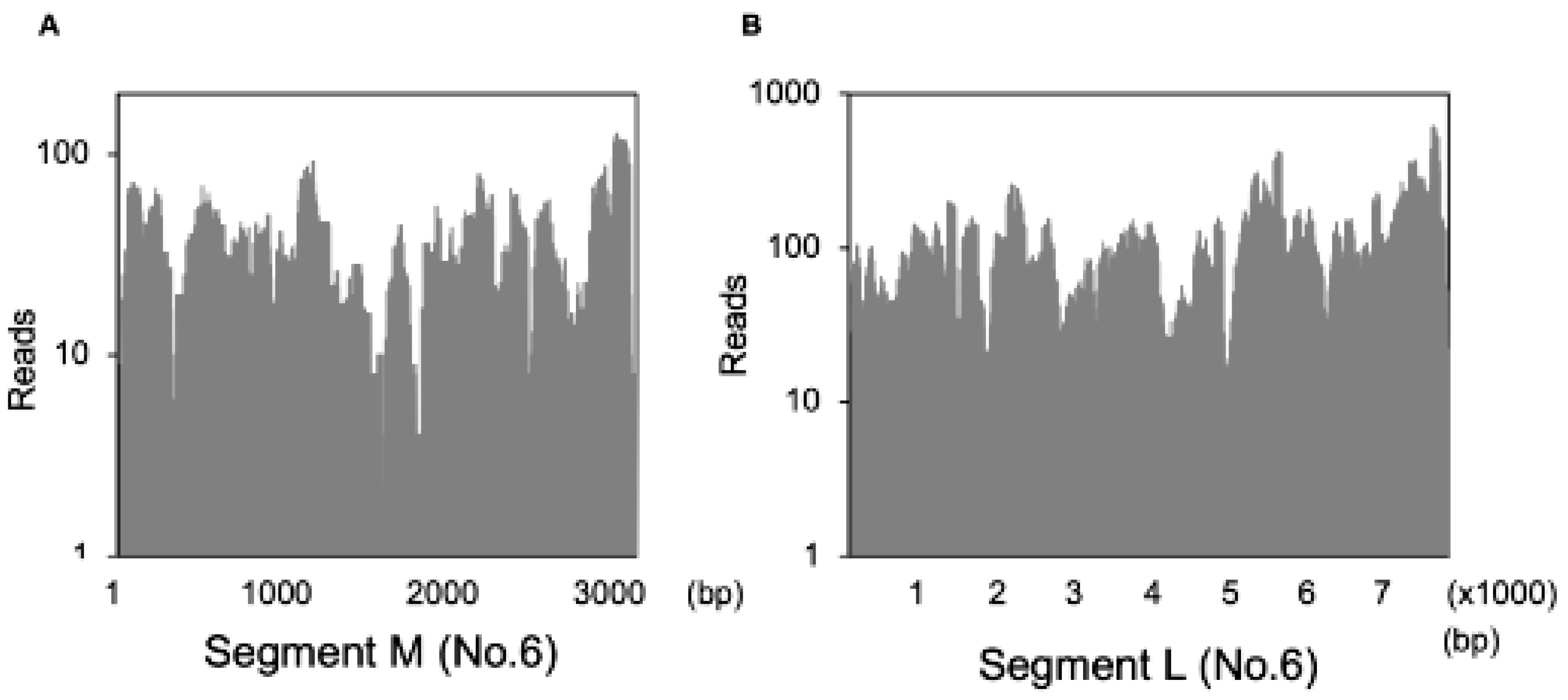
3.6. Detection of the Fulton Virus in Wild Mus Musculus
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koonin, E.V. The two empires and three domains of life in the postgenomic age. Nat. Educ. 2010, 3, 27. [Google Scholar]
- Roossinck, M.J. The good viruses: Viral mutualistic symbioses. Nat. Rev. Microbiol. 2011, 9, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Bell, B.P.; Damon, I.K. Overview, control strategies, and lessons learned in the CDC response to the 2014–2016 ebola epidemic. MMWR Suppl. 2016, 65, 4–11. [Google Scholar] [CrossRef] [Green Version]
- Languon, S.; Quaye, O. Filovirus disease outbreaks: A chronological overview. Virology 2019, 10, 1–12. [Google Scholar] [CrossRef]
- Lipkin, W.I. The changing face of pathogen discovery and surveillance. Nat. Rev. Microbiol. 2013, 11, 133–141. [Google Scholar] [CrossRef]
- Hiscox, J.A. RNA viruses: Hijacking the dynamics nucleolus. Nat. Rev. Microbiol. 2007, 5, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Al Rwahnih, M.; Daubert, S. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef] [Green Version]
- Coetzee, B.; Freeborough, M.J. Deep sequencing analysis of viruses infecting grapevines: Virome of a vineyard. Virology 2010, 400, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decker, C.J.; Parker, R. Analysis of double-stranded RNA from microbial communities identifies double-stranded RNA virus-like elements. Cell Rep. 2014, 7, 898–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradpour, D.; Penin, F. Replication of hepatitis C virus. Nat. Rev. Microbiol. 2007, 5, 453–463. [Google Scholar] [CrossRef]
- Te Velthuis, A.J.; Fodor, E. Influenza virus RNA polymerase: Insights into the mechanisms of viral RNA synthesis. Nat. Rev. Microbiol. 2016, 14, 479–493. [Google Scholar] [CrossRef] [Green Version]
- Yanagisawa, H.; Tomita, R. Combined DECS analysis and next-generation sequencing enable efficient detection of novel plant RNA viruses. Viruses 2016, 8, 70. [Google Scholar] [CrossRef] [Green Version]
- Nerva, L.; Ciuffo, M. Multiple approaches for the detection and characterization of viral and plasmid symbionts from a collection of marine fungi. Virus Res. 2016, 219, 22–38. [Google Scholar] [CrossRef]
- Urayama, S.; Takaki, Y. Unveiling the RNA virosphere associated with marine microorganisms. Mol. Ecol. Resour. 2018, 18, 1444–1455. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.R.; Lake, J.R. OPTN/SRTR 2017 annual date repot: Liver. Am. J. Transplant. 2019, 19, 184–283. [Google Scholar] [CrossRef] [Green Version]
- Kanai, Y.; Kawagishi, T. Lethal murine infection model for human respiratory disease-associated Pteropine orthoreovirus. Virology 2018, 514, 57–65. [Google Scholar] [CrossRef]
- Kawagishi, T.; Kanai, Y. Reverse genetics for fusogenic bat-borne orthoreovirus associated with acute respiratory tract infection in humans: Role of outer capsid protein σc in viral replication and pathogens. PLoS Pathog. 2016, 12, e1005455. [Google Scholar] [CrossRef] [Green Version]
- Nanahara, M.; Chang, Y.T. HBV Pre-S1-Derived Myristoylated Peptide (Myr47): Identification of the Inhibitory Activity on the Cellular Uptake of Lipid Nanoparticles. Viruses 2021, 13, 929. [Google Scholar] [CrossRef]
- Okada, R.; Kiyota, E. A simple and rapid method to purify viral dsRNA from plant and fungal tissue. J. Gen. Plant. Pathol. 2015, 81, 103–107. [Google Scholar] [CrossRef]
- Urayama, S.; Takashima, Y. A new fractionation and recovery method of viral genomes based on nucleic acid composition and structure using tandem column chromatography. Microbes Environ. 2015, 30, 199–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masaki, T.; Suzuki, R. Production of infectious hepatitis C virus by using RNA polymerase I-mediated transcription. J. Virol. 2010, 84, 5824–5835. [Google Scholar] [CrossRef] [Green Version]
- Wakita, T.; Pietschmann, T. Production infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [Green Version]
- Russel, R.S.; Meunier, J.C. Advantage of a single-cycle production assay to study cell culture-adaptive mutations of hepatitis C virus. Proc. Natl. Acad. Sci. USA 2008, 105, 4370–4375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Zhong, G. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049.28. [Google Scholar] [CrossRef] [PubMed]
- Watashi, K.; Sluder, A. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP). Hepatology 2014, 59, 1726–1737. [Google Scholar] [CrossRef]
- Qi, L.S.; Larson, M.H. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Nassal, M.; Rieger, A. A bulged region of the hepatitis B virus RNA encapsidation signal contains the replication origin for discontinuous first-stranded DNA synthesis. J. Virol. 1996, 70, 2764–2773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, S.H.; Che, X. Discovery of two highly divergent negative-sense RNA viruses associated with the parasitic nematode, Capillaria hepatica, in wild Mus musculus from New York City. J. Gen. Virol. 2020, 100, 1350–1362. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Disease | No. | Disease |
|---|---|---|---|
| 1 | HCC (HCV) | 12 | Wilson disease |
| 2 | LC (HCV) | PBC | |
| 3 | HCC (HCV) | AIH | |
| HCC (HCV) | LC (HBV), HCC | ||
| 4 | NASH | 13 | AIH |
| NASH, HCC | NASH | ||
| 5 | AIH, PBC | LC (HCV) | |
| AIH, PBC | NASH | ||
| 6 | Calori disease | 14 | LC (Alcohol) |
| PSC | LC (Alcohol), HCC | ||
| 7 | VOD | NASH | |
| LC (Alcohol) | LC (HBV), HCC | ||
| Wilson disease | 15 | LC, HCC | |
| LC (nonBnonC) | LC (Alcohol) | ||
| 8 | LC (HCV) | NASH | |
| LC (nonBnonC) | 16 | Acute liver failure | |
| PBC | LC (Alcohol) | ||
| HCC (HBV) | PBC | ||
| 9 | LC (Alcohol) | 17 | LC (HCV) |
| Acute liver failure (HBV) | HBV | ||
| LC (Alcohol) | PBC | ||
| AIH, HCC | 18 | Acute liver failure (HCV) | |
| 10 | Acute liver failure (HBV) | LC (Alcohol) | |
| LC (nonBnonC), HCC | LC (HBV) | ||
| LC (HCV) | 19 | LC (HCV) | |
| PBC | AIH, PSC | ||
| 11 | LC (nonBnonC), HCC | LC (Alcohol) | |
| LC (HBV) | |||
| LC (Alcohol) | |||
| LC (HCV), HCC |
| Total Reads | Host | Bacteria | Viruses | Fungi | Others | Unassigned | |
|---|---|---|---|---|---|---|---|
| Total RNA-seq | |||||||
| Fungus | 488,608 | 14,119 (2.9%) | 28,056 (5.7%) | 22,115 (4.5%) | 216,259 (44.3%) | 956 (0.2%) | 207,103 (42.4%) |
| Liver (HCV1) | 1,121,844 | 915,137 (81.6%) | 166 (0.0%) | 247 (0.0%) | 8862 (0.8%) | 4482 (0.4%) | 192,950 (17.2%) |
| Liver (HCV2) | 734,937 | 624,222 (84.9%) | 610 (0.1%) | 73 (0.0%) | 5275 (0.7%) | 2752 (0.4%) | 102,005 (13.9%) |
| FLDS | |||||||
| Fungus | 812,459 | 553 (0.1%) | 44 (0.0%) | 447,923 (55.1%) | 30,944 (3.8%) | 586 (0.1%) | 332,409 (40.9%) |
| Liver (HCV1) | 235,899 | 35,165 (14.9%) | 376 (0.2%) | 214 (0.1%) | 76,749 (32.5%) | 1071 (0.5%) | 122,324 (51.9%) |
| Liver (HCV2) | 338,312 | 33,828 (10.0%) | 743 (0.2%) | 229 (0.1%) | 108,961 (32.2%) | 2588 (0.8%) | 191,963 (56.7%) |
| Modified dsRNA-seq | |||||||
| Fungus | 926,151 | 67 (0.0%) | 31 (0.0%) | 242,514 (26.2%) | 12 (0.0%) | 2 (0.0%) | 683,525 (73.8%) |
| Liver (HCV1) | 717,978 | 452,549 (63.0%) | 12,419 (1.7%) | 2953 (0.4%) | 358 (0.0%) | 93 (0.0%) | 249,606 (34.8%) |
| Liver (HCV2) | 635,743 | 486,372 (76.5%) | 3264 (0.5%) | 2464 (0.4%) | 196 (0.0%) | 117 (0.0%) | 143,330 (22.5%) |
| No. | Total | Viruses | Ratio | Virus (Reads) | |
|---|---|---|---|---|---|
| (Reads) | (Reads) | (%) | Top Hit | ||
| 1 | 717,978 | 2953 | 0.4 | Hepatitis C virus | 2889 |
| 2 | 635,743 | 2464 | 0.4 | Hepatitis C virus | 2430 |
| 3 | 556,921 | 10,956 | 2.0 | Brome mosaic virus | 8927 |
| Hepatitis C virus | 2011 | ||||
| 4 | 609,020 | 43 | 0.0 | Human endogenous retrovirus K | 35 |
| 5 | 595,735 | 13 | 0.0 | Human endogenous retrovirus K | 12 |
| 6 | 563,170 | 38 | 0.0 | Human endogenous retrovirus K | 32 |
| 7 | 17,634,237 | 960 | 0.0 | Human endogenous retrovirus K | 482 |
| 8 | 15,890,244 | 2659 | 0.0 | Human endogenous retrovirus K | 2081 |
| 9 | 17,626,711 | 1091 | 0.0 | Human endogenous retrovirus K | 1064 |
| 10 | 12,808,763 | 341,827 | 2.7 | Hepatitis C virus | 341,738 |
| 11 | 14,061,933 | 220,722 | 1.6 | Hepatitis C virus | 195,642 |
| Hepatitis B virus | 24,960 | ||||
| 12 | 18,478,904 | 636 | 0.0 | Bell pepper endornavirus | 272 |
| 13 | 16,294,029 | 8303 | 0.1 | Hepatitis C virus | 7298 |
| 14 | 16,097,692 | 1085 | 0.0 | Human endogenous retrovirus K | 1076 |
| 15 | 12,106,817 | 1273 | 0.0 | Human endogenous retrovirus K | 568 |
| 16 | 15,198,287 | 764 | 0.0 | Human endogenous retrovirus K | 392 |
| 17 | 20,526,142 | 3935 | 0.0 | Hepatitis C virus | 2628 |
| 18 | 15,195,371 | 2641 | 0.0 | Hepatitis C virus | 2347 |
| 19 | 19,094,227 | 1260 | 0.0 | Human endogenous retrovirus K | 862 |
| No. | Type of Wild Rodents | Sample | Organ | Identification |
|---|---|---|---|---|
| 1 | Apodemus speciosus | 5 | liver | |
| 2 | Myodes rufocanus bedfordiae | 5 | liver | |
| 3 | Myodes rufocanus bedfordiae | 5 | liver | Fulton virus (L, M) |
| 4 | Myodes rufocanus bedfordiae | 5 | liver | Fulton virus (L, M) |
| 5 | Myodes rufocanus bedfordiae | 5 | liver | |
| 6 | Myodes rufocanus bedfordiae | 5 | liver | Fulton virus (L, M) |
| 7 | Myodes rufocanus bedfordiae | 5 | liver | |
| 8 | Myodes rufocanus bedfordiae | 5 | liver | |
| 9 | Myodes rufocanus bedfordiae | 4 | liver |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izumi, T.; Morioka, Y.; Urayama, S.-i.; Motooka, D.; Tamura, T.; Kawagishi, T.; Kanai, Y.; Kobayashi, T.; Ono, C.; Morinaga, A.; et al. DsRNA Sequencing for RNA Virus Surveillance Using Human Clinical Samples. Viruses 2021, 13, 1310. https://doi.org/10.3390/v13071310
Izumi T, Morioka Y, Urayama S-i, Motooka D, Tamura T, Kawagishi T, Kanai Y, Kobayashi T, Ono C, Morinaga A, et al. DsRNA Sequencing for RNA Virus Surveillance Using Human Clinical Samples. Viruses. 2021; 13(7):1310. https://doi.org/10.3390/v13071310
Chicago/Turabian StyleIzumi, Takuma, Yuhei Morioka, Syun-ichi Urayama, Daisuke Motooka, Tomokazu Tamura, Takahiro Kawagishi, Yuta Kanai, Takeshi Kobayashi, Chikako Ono, Akinari Morinaga, and et al. 2021. "DsRNA Sequencing for RNA Virus Surveillance Using Human Clinical Samples" Viruses 13, no. 7: 1310. https://doi.org/10.3390/v13071310
APA StyleIzumi, T., Morioka, Y., Urayama, S. -i., Motooka, D., Tamura, T., Kawagishi, T., Kanai, Y., Kobayashi, T., Ono, C., Morinaga, A., Tomiyama, T., Iseda, N., Kosai, Y., Inokuchi, S., Nakamura, S., Tanaka, T., Moriishi, K., Kariwa, H., Yoshizumi, T., ... Fukuhara, T. (2021). DsRNA Sequencing for RNA Virus Surveillance Using Human Clinical Samples. Viruses, 13(7), 1310. https://doi.org/10.3390/v13071310