
Mechanism of Hepatitis B Virus cccDNA Formation


Abstract
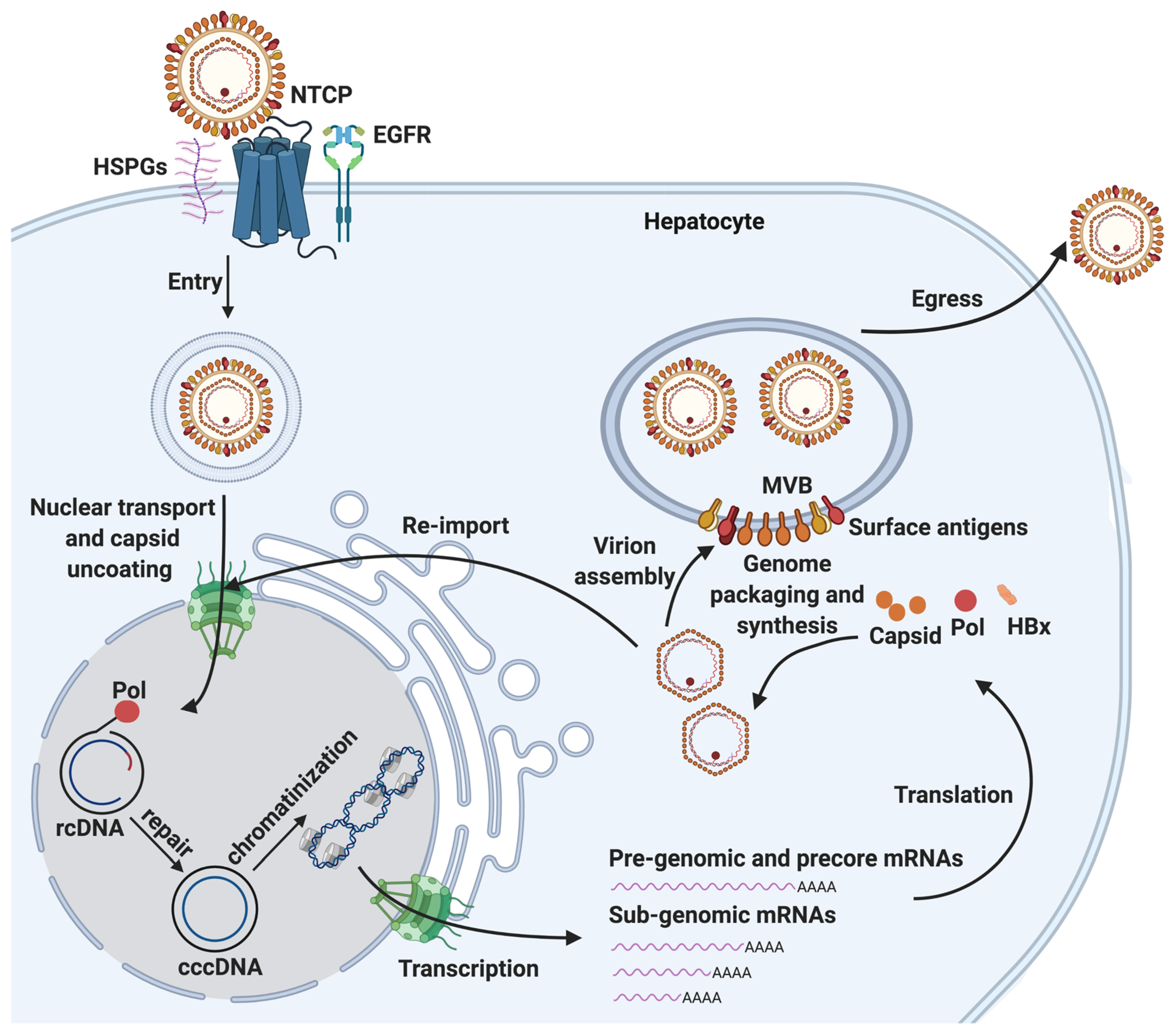
:1. Overview of HBV Life Cycle and cccDNA Biogenesis
2. Functions of HBV Viral Factors in rcDNA Repair
3. General Approaches and Challenges of Studying cccDNA Formation
4. General Steps Involved in rcDNA Repair
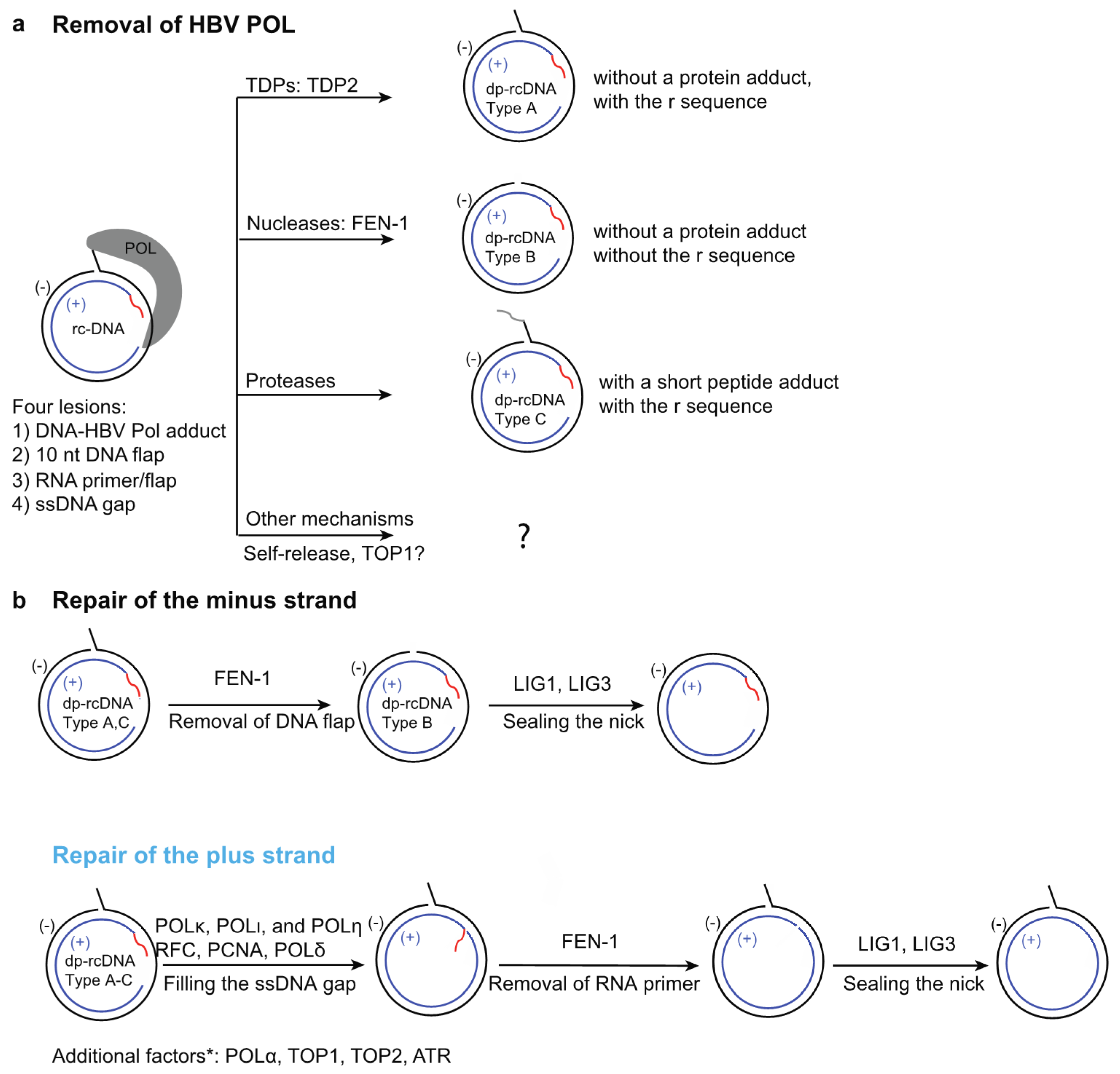
4.1. Removal of HBV POL from HBV rcDNA
4.1.1. POL Removal—Release by Tyrosyl-DNA Phosphodiesterases (TDPs)
4.1.2. POL Removal—Release by FEN-1 Endonuclease
4.1.3. POL Removal—Release by Proteases
4.1.4. POL Removal—Additional Release Mechanisms
4.2. Removal of the Terminal-Redundancy Sequence r
4.3. RNA Removal of HBV rcDNA
4.4. Completion of Synthesis of the Plus Strand
4.5. Ligation of Nicks on Both Strands
5. DNA Damage Response and HBV rcDNA Repair
6. Differences in cccDNA Formation of HBV and DHBV
7. cccDNA Biogenesis in Murine Cells
8. Targeting DNA Repair Machinery as a Potential Treatment for HBV Infection
9. Concluding Remarks
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lang, J.; Neumann-Haefelin, C.; Thimme, R. Immunological cure of HBV infection. Hepatol. Int. 2019, 13, 113–124. [Google Scholar] [CrossRef]
- Yuen, M.F.; Chen, D.S.; Dusheiko, G.M.; Janssen, H.L.A.; Lau, D.T.Y.; Locarnini, S.A.; Peters, M.G.; Lai, C.L. Hepatitis B virus infection. Nat. Rev. Dis. Primers 2018, 4, 18035. [Google Scholar] [CrossRef]
- Schweitzer, A.; Horn, J.; Mikolajczyk, R.T.; Krause, G.; Ott, J.J. Estimations of worldwide prevalence of chronic hepatitis B virus infection: A systematic review of data published between 1965 and 2013. Lancet 2015, 386, 1546–1555. [Google Scholar] [CrossRef]
- Revill, P.A.; Chisari, F.V.; Block, J.M.; Dandri, M.; Gehring, A.J.; Guo, H.; Hu, J.; Kramvis, A.; Lampertico, P.; Janssen, H.L.A.; et al. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol. Hepatol. 2019, 4, 545–558. [Google Scholar] [CrossRef]
- Mason, W.S. Animal models and the molecular biology of hepadnavirus infection. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Will, H.; Cattaneo, R.; Koch, H.G.; Darai, G.; Schaller, H.; Schellekens, H.; van Eerd, P.M.; Deinhardt, F. Cloned HBV DNA causes hepatitis in chimpanzees. Nature 1982, 299, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Winer, B.Y.; Ploss, A. Determinants of hepatitis B and delta virus host tropism. Curr. Opin. Virol. 2015, 13, 109–116. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Maya, S.; Ploss, A. Animal Models of Hepatitis B Virus Infection-Success, Challenges, and Future Directions. Viruses 2021, 13, 777. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, S. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World J. Gastroenterol. 2007, 13, 14–21. [Google Scholar] [CrossRef] [Green Version]
- Suh, A.; Brosius, J.; Schmitz, J.; Kriegs, J.O. The genome of a Mesozoic paleovirus reveals the evolution of hepatitis B viruses. Nat. Commun. 2013, 4, 1791. [Google Scholar] [CrossRef] [Green Version]
- Drexler, J.F.; Geipel, A.; Konig, A.; Corman, V.M.; van Riel, D.; Leijten, L.M.; Bremer, C.M.; Rasche, A.; Cottontail, V.M.; Maganga, G.D.; et al. Bats carry pathogenic hepadnaviruses antigenically related to hepatitis B virus and capable of infecting human hepatocytes. Proc. Natl. Acad. Sci. USA 2013, 110, 16151–16156. [Google Scholar] [CrossRef] [Green Version]
- Suh, A.; Weber, C.C.; Kehlmaier, C.; Braun, E.L.; Green, R.E.; Fritz, U.; Ray, D.A.; Ellegren, H. Early mesozoic coexistence of amniotes and hepadnaviridae. PLoS Genet. 2014, 10, e1004559. [Google Scholar] [CrossRef] [Green Version]
- Dill, J.A.; Camus, A.C.; Leary, J.H.; Di Giallonardo, F.; Holmes, E.C.; Ng, T.F. Distinct Viral Lineages from Fish and Amphibians Reveal the Complex Evolutionary History of Hepadnaviruses. J. Virol. 2016, 90, 7920–7933. [Google Scholar] [CrossRef] [Green Version]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Borold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe 2017, 22, 387–399.e6. [Google Scholar] [CrossRef] [Green Version]
- Nassal, M. HBV cccDNA: Viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut 2015, 64, 1972–1984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schultz, U.; Grgacic, E.; Nassal, M. Duck hepatitis B virus: An invaluable model system for HBV infection. Adv. Virus Res. 2004, 63, 1–70. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Schaller, H. Hepadnaviral assembly is initiated by polymerase binding to the encapsidation signal in the viral RNA genome. EMBO J. 1992, 11, 3413–3420. [Google Scholar] [CrossRef]
- Hirsch, R.C.; Loeb, D.D.; Pollack, J.R.; Ganem, D. cis-acting sequences required for encapsidation of duck hepatitis B virus pregenomic RNA. J. Virol. 1991, 65, 3309–3316. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Ploss, A. Hepatitis B virus cccDNA is formed through distinct repair processes of each strand. Nat. Commun. 2021, 12, 1591. [Google Scholar] [CrossRef]
- Selzer, L.; Zlotnick, A. Assembly and Release of Hepatitis B Virus. Cold Spring Harb. Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Zlotnick, A.; Venkatakrishnan, B.; Tan, Z.; Lewellyn, E.; Turner, W.; Francis, S. Core protein: A pleiotropic keystone in the HBV lifecycle. Antiviral Res. 2015, 121, 82–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, T.; Tanaka, Y. The Role of Hepatitis B Core-Related Antigen. Genes 2019, 10, 357. [Google Scholar] [CrossRef] [Green Version]
- Bouchard, M.J.; Schneider, R.J. The enigmatic X gene of hepatitis B virus. J. Virol. 2004, 78, 12725–12734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benhenda, S.; Cougot, D.; Buendia, M.A.; Neuveut, C. Hepatitis B virus X protein molecular functions and its role in virus life cycle and pathogenesis. Adv. Cancer Res. 2009, 103, 75–109. [Google Scholar] [CrossRef]
- Slagle, B.L.; Bouchard, M.J. Role of HBx in hepatitis B virus persistence and its therapeutic implications. Curr. Opin. Virol. 2018, 30, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Belloni, L.; Pollicino, T.; De Nicola, F.; Guerrieri, F.; Raffa, G.; Fanciulli, M.; Raimondo, G.; Levrero, M. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA 2009, 106, 19975–19979. [Google Scholar] [CrossRef] [Green Version]
- Schulze, A.; Gripon, P.; Urban, S. Hepatitis B virus infection initiates with a large surface protein-dependent binding to heparan sulfate proteoglycans. Hepatology 2007, 46, 1759–1768. [Google Scholar] [CrossRef]
- Verrier, E.R.; Colpitts, C.C.; Bach, C.; Heydmann, L.; Weiss, A.; Renaud, M.; Durand, S.C.; Habersetzer, F.; Durantel, D.; Abou-Jaoude, G.; et al. A targeted functional RNA interference screen uncovers glypican 5 as an entry factor for hepatitis B and D viruses. Hepatology 2016, 63, 35–48. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef]
- Ni, Y.; Lempp, F.A.; Mehrle, S.; Nkongolo, S.; Kaufman, C.; Falth, M.; Stindt, J.; Koniger, C.; Nassal, M.; Kubitz, R.; et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 2014, 146, 1070–1083. [Google Scholar] [CrossRef]
- Iwamoto, M.; Saso, W.; Sugiyama, R.; Ishii, K.; Ohki, M.; Nagamori, S.; Suzuki, R.; Aizaki, H.; Ryo, A.; Yun, J.H.; et al. Epidermal growth factor receptor is a host-entry cofactor triggering hepatitis B virus internalization. Proc. Natl. Acad. Sci. USA 2019, 116, 8487–8492. [Google Scholar] [CrossRef] [Green Version]
- Iwamoto, M.; Saso, W.; Nishioka, K.; Ohashi, H.; Sugiyama, R.; Ryo, A.; Ohki, M.; Yun, J.H.; Park, S.Y.; Ohshima, T.; et al. The machinery for endocytosis of epidermal growth factor receptor coordinates the transport of incoming hepatitis B virus to the endosomal network. J. Biol. Chem. 2020, 295, 800–807. [Google Scholar] [CrossRef]
- Tang, L.; Sheraz, M.; McGrane, M.; Chang, J.; Guo, J.T. DNA Polymerase alpha is essential for intracellular amplification of hepatitis B virus covalently closed circular DNA. PLoS Pathog. 2019, 15, e1007742. [Google Scholar] [CrossRef] [PubMed]
- Li, H.C.; Huang, E.Y.; Su, P.Y.; Wu, S.Y.; Yang, C.C.; Lin, Y.S.; Chang, W.C.; Shih, C. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog. 2010, 6, e1001162. [Google Scholar] [CrossRef] [Green Version]
- Kann, M.; Sodeik, B.; Vlachou, A.; Gerlich, W.H.; Helenius, A. Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J. Cell Biol. 1999, 145, 45–55. [Google Scholar] [CrossRef]
- Rabe, B.; Vlachou, A.; Pante, N.; Helenius, A.; Kann, M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc. Natl. Acad. Sci. USA 2003, 100, 9849–9854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, C.T.; Liaw, Y.F.; Ou, J.H. The arginine-rich domain of hepatitis B virus precore and core proteins contains a signal for nuclear transport. J. Virol. 1990, 64, 6141–6147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Mao, R.; Block, T.M.; Guo, J.T. Production and function of the cytoplasmic deproteinized relaxed circular DNA of hepadnaviruses. J. Virol. 2010, 84, 387–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nair, S.; Zlotnick, A. HBV Core Protein Is in Flux between Cytoplasmic, Nuclear, and Nucleolar Compartments. mBio 2021, 12. [Google Scholar] [CrossRef]
- Luo, J.; Xi, J.; Gao, L.; Hu, J. Role of Hepatitis B virus capsid phosphorylation in nucleocapsid disassembly and covalently closed circular DNA formation. PLoS Pathog. 2020, 16, e1008459. [Google Scholar] [CrossRef] [Green Version]
- Lupberger, J.; Schaedler, S.; Peiran, A.; Hildt, E. Identification and characterization of a novel bipartite nuclear localization signal in the hepatitis B virus polymerase. World J. Gastroenterol. 2013, 19, 8000–8010. [Google Scholar] [CrossRef]
- Bock, C.T.; Schranz, P.; Schroder, C.H.; Zentgraf, H. Hepatitis B virus genome is organized into nucleosomes in the nucleus of the infected cell. Virus Genes 1994, 8, 215–229. [Google Scholar] [CrossRef]
- Bock, C.T.; Schwinn, S.; Locarnini, S.; Fyfe, J.; Manns, M.P.; Trautwein, C.; Zentgraf, H. Structural organization of the hepatitis B virus minichromosome. J. Mol. Biol. 2001, 307, 183–196. [Google Scholar] [CrossRef]
- Tropberger, P.; Mercier, A.; Robinson, M.; Zhong, W.; Ganem, D.E.; Holdorf, M. Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA 2015, 112, E5715–E5724. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Seeger, C. Hepadnavirus Genome Replication and Persistence. Cold Spring Harb. Perspect. Med. 2015, 5, a021386. [Google Scholar] [CrossRef] [Green Version]
- Beck, J.; Nassal, M. Hepatitis B virus replication. World J. Gastroenterol. 2007, 13, 48–64. [Google Scholar] [CrossRef] [Green Version]
- Nassal, M. Hepatitis B viruses: Reverse transcription a different way. Virus Res. 2008, 134, 235–249. [Google Scholar] [CrossRef]
- Hu, J.; Tang, L.; Cheng, J.; Zhou, T.; Li, Y.; Chang, J.; Zhao, Q.; Guo, J.T. Hepatitis B virus nucleocapsid uncoating: Biological consequences and regulation by cellular nucleases. Emerg. Microbes Infect. 2021, 10, 852–864. [Google Scholar] [CrossRef]
- Lucifora, J.; Protzer, U. Attacking hepatitis B virus cccDNA—The holy grail to hepatitis B cure. J. Hepatol. 2016, 64, S41–S48. [Google Scholar] [CrossRef]
- Long, Q.; Yan, R.; Hu, J.; Cai, D.; Mitra, B.; Kim, E.S.; Marchetti, A.; Zhang, H.; Wang, S.; Liu, Y.; et al. The role of host DNA ligases in hepadnavirus covalently closed circular DNA formation. PLoS Pathog. 2017, 13, e1006784. [Google Scholar] [CrossRef]
- Seeger, C.; Mason, W.S. Molecular biology of hepatitis B virus infection. Virology 2015, 479–480, 672–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, Y.; Guo, H. Hepatitis B virus cccDNA: Formation, regulation and therapeutic potential. Antivir. Res. 2020, 180, 104824. [Google Scholar] [CrossRef] [PubMed]
- Marchetti, A.L.; Guo, H. New Insights on Molecular Mechanism of Hepatitis B Virus Covalently Closed Circular DNA Formation. Cells 2020, 9, 2430. [Google Scholar] [CrossRef]
- Jiang, B.; Hildt, E. Intracellular Trafficking of HBV Particles. Cells 2020, 9, 2023. [Google Scholar] [CrossRef]
- Kann, M.; Schmitz, A.; Rabe, B. Intracellular transport of hepatitis B virus. World J. Gastroenterol. 2007, 13, 39–47. [Google Scholar] [CrossRef]
- Diogo Dias, J.; Sarica, N.; Neuveut, C. Early Steps of Hepatitis B Life Cycle: From Capsid Nuclear Import to cccDNA Formation. Viruses 2021, 13, 757. [Google Scholar] [CrossRef]
- Tsukuda, S.; Watashi, K. Hepatitis B virus biology and life cycle. Antivir. Res. 2020, 182, 104925. [Google Scholar] [CrossRef]
- Kaplan, P.M.; Greenman, R.L.; Gerin, J.L.; Purcell, R.H.; Robinson, W.S. DNA polymerase associated with human hepatitis B antigen. J. Virol. 1973, 12, 995–1005. [Google Scholar] [CrossRef] [Green Version]
- Robinson, W.S.; Clayton, D.A.; Greenman, R.L. DNA of a human hepatitis B virus candidate. J. Virol. 1974, 14, 384–391. [Google Scholar] [CrossRef] [Green Version]
- Robinson, W.S.; Greenman, R.L. DNA polymerase in the core of the human hepatitis B virus candidate. J. Virol. 1974, 13, 1231–1236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hantz, O.; Parent, R.; Durantel, D.; Gripon, P.; Guguen-Guillouzo, C.; Zoulim, F. Persistence of the hepatitis B virus covalently closed circular DNA in HepaRG human hepatocyte-like cells. J. Gen. Virol. 2009, 90, 127–135. [Google Scholar] [CrossRef]
- Qi, Y.; Gao, Z.; Xu, G.; Peng, B.; Liu, C.; Yan, H.; Yao, Q.; Sun, G.; Liu, Y.; Tang, D.; et al. DNA Polymerase kappa Is a Key Cellular Factor for the Formation of Covalently Closed Circular DNA of Hepatitis B Virus. PLoS Pathog. 2016, 12, e1005893. [Google Scholar] [CrossRef] [Green Version]
- Xia, Y.; Carpentier, A.; Cheng, X.; Block, P.D.; Zhao, Y.; Zhang, Z.; Protzer, U.; Liang, T.J. Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J. Hepatol. 2017, 66, 494–503. [Google Scholar] [CrossRef] [Green Version]
- Kock, J.; Schlicht, H.J. Analysis of the earliest steps of hepadnavirus replication: Genome repair after infectious entry into hepatocytes does not depend on viral polymerase activity. J. Virol. 1993, 67, 4867–4874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraleda, G.; Saputelli, J.; Aldrich, C.E.; Averett, D.; Condreay, L.; Mason, W.S. Lack of effect of antiviral therapy in nondividing hepatocyte cultures on the closed circular DNA of woodchuck hepatitis virus. J. Virol. 1997, 71, 9392–9399. [Google Scholar] [CrossRef] [Green Version]
- Lenhoff, R.J.; Summers, J. Coordinate regulation of replication and virus assembly by the large envelope protein of an avian hepadnavirus. J. Virol. 1994, 68, 4565–4571. [Google Scholar] [CrossRef] [Green Version]
- Summers, J.; Smith, P.M.; Horwich, A.L. Hepadnavirus envelope proteins regulate covalently closed circular DNA amplification. J. Virol. 1990, 64, 2819–2824. [Google Scholar] [CrossRef] [Green Version]
- Kock, J.; Rosler, C.; Zhang, J.J.; Blum, H.E.; Nassal, M.; Thoma, C. Generation of covalently closed circular DNA of hepatitis B viruses via intracellular recycling is regulated in a virus specific manner. PLoS Pathog. 2010, 6, e1001082. [Google Scholar] [CrossRef]
- Lentz, T.B.; Loeb, D.D. Roles of the envelope proteins in the amplification of covalently closed circular DNA and completion of synthesis of the plus-strand DNA in hepatitis B virus. J. Virol. 2011, 85, 11916–11927. [Google Scholar] [CrossRef] [Green Version]
- Tu, T.; Zehnder, B.; Qu, B.; Urban, S. De novo synthesis of hepatitis B virus nucleocapsids is dispensable for the maintenance and transcriptional regulation of cccDNA. JHEP Rep. 2021, 3, 100195. [Google Scholar] [CrossRef]
- Decorsiere, A.; Mueller, H.; van Breugel, P.C.; Abdul, F.; Gerossier, L.; Beran, R.K.; Livingston, C.M.; Niu, C.; Fletcher, S.P.; Hantz, O.; et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature 2016, 531, 386–389. [Google Scholar] [CrossRef]
- Schreiner, S.; Nassal, M. A Role for the Host DNA Damage Response in Hepatitis B Virus cccDNA Formation-and Beyond? Viruses 2017, 9, 125. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.T.; Pryce, M.; Wang, X.; Barrasa, M.I.; Hu, J.; Seeger, C. Conditional replication of duck hepatitis B virus in hepatoma cells. J. Virol. 2003, 77, 1885–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladner, S.K.; Otto, M.J.; Barker, C.S.; Zaifert, K.; Wang, G.H.; Guo, J.T.; Seeger, C.; King, R.W. Inducible expression of human hepatitis B virus (HBV) in stably transfected hepatoblastoma cells: A novel system for screening potential inhibitors of HBV replication. Antimicrob. Agents Chemother. 1997, 41, 1715–1720. [Google Scholar] [CrossRef] [Green Version]
- Wei, L.; Ploss, A. Core components of DNA lagging strand synthesis machinery are essential for hepatitis B virus cccDNA formation. Nat. Microbiol. 2020, 5, 715–726. [Google Scholar] [CrossRef]
- Koniger, C.; Wingert, I.; Marsmann, M.; Rosler, C.; Beck, J.; Nassal, M. Involvement of the host DNA-repair enzyme TDP2 in formation of the covalently closed circular DNA persistence reservoir of hepatitis B viruses. Proc. Natl. Acad. Sci. USA 2014, 111, E4244–E4253. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, K.; Que, L.; Shimadu, M.; Koura, M.; Ishihara, Y.; Wakae, K.; Nakamura, T.; Watashi, K.; Wakita, T.; Muramatsu, M. Flap endonuclease 1 is involved in cccDNA formation in the hepatitis B virus. PLoS Pathog. 2018, 14, e1007124. [Google Scholar] [CrossRef] [PubMed]
- Gerlich, W.H.; Robinson, W.S. Hepatitis B virus contains protein attached to the 5’ terminus of its complete DNA strand. Cell 1980, 21, 801–809. [Google Scholar] [CrossRef]
- Bosch, V.; Bartenschlager, R.; Radziwill, G.; Schaller, H. The duck hepatitis B virus P-gene codes for protein strongly associated with the 5’-end of the viral DNA minus strand. Virology 1988, 166, 475–485. [Google Scholar] [CrossRef]
- Gong, Y.; Yao, E.; Tavis, J.E. Evidence that the RNAseH activity of the duck hepatitis B virus is unable to act on exogenous substrates. BMC Microbiol. 2001, 1, 12. [Google Scholar] [CrossRef] [PubMed]
- Radziwill, G.; Zentgraf, H.; Schaller, H.; Bosch, V. The duck hepatitis B virus DNA polymerase is tightly associated with the viral core structure and unable to switch to an exogenous template. Virology 1988, 163, 123–132. [Google Scholar] [CrossRef]
- Gao, W.; Hu, J. Formation of hepatitis B virus covalently closed circular DNA: Removal of genome-linked protein. J. Virol. 2007, 81, 6164–6174. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Jiang, D.; Zhou, T.; Cuconati, A.; Block, T.M.; Guo, J.T. Characterization of the intracellular deproteinized relaxed circular DNA of hepatitis B virus: An intermediate of covalently closed circular DNA formation. J. Virol. 2007, 81, 12472–12484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, J.; Cui, X.; Gao, L.; Hu, J. Identification of Intermediate in Hepatitis B Virus CCC DNA Formation and Sensitive and Selective CCC DNA Detection. J. Virol. 2017, 91, e00539-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ko, C.; Chakraborty, A.; Chou, W.M.; Hasreiter, J.; Wettengel, J.M.; Stadler, D.; Bester, R.; Asen, T.; Zhang, K.; Wisskirchen, K.; et al. Hepatitis B virus genome recycling and de novo secondary infection events maintain stable cccDNA levels. J. Hepatol. 2018, 69, 1231–1241. [Google Scholar] [CrossRef]
- Dezhbord, M.; Lee, S.; Kim, W.; Seong, B.L.; Ryu, W.S. Characterization of the molecular events of covalently closed circular DNA synthesis in de novo Hepatitis B virus infection of human hepatoma cells. Antivir. Res. 2019, 163, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Rumin, S.; Urban, S.; Le Seyec, J.; Glaise, D.; Cannie, I.; Guyomard, C.; Lucas, J.; Trepo, C.; Guguen-Guillouzo, C. Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 2002, 99, 15655–15660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, D.; Yan, R.; Xu, J.Z.; Zhang, H.; Shen, S.; Mitra, B.; Marchetti, A.; Kim, E.S.; Guo, H. Characterization of the Termini of Cytoplasmic Hepatitis B Virus Deproteinated Relaxed Circular DNA. J. Virol. 2020, 95, e00922-20. [Google Scholar] [CrossRef]
- Miller, R.H.; Robinson, W.S. Hepatitis B virus DNA forms in nuclear and cytoplasmic fractions of infected human liver. Virology 1984, 137, 390–399. [Google Scholar] [CrossRef]
- Kawale, A.S.; Povirk, L.F. Tyrosyl-DNA phosphodiesterases: Rescuing the genome from the risks of relaxation. Nucleic Acids Res. 2018, 46, 520–537. [Google Scholar] [CrossRef] [Green Version]
- Cortes Ledesma, F.; El Khamisy, S.F.; Zuma, M.C.; Osborn, K.; Caldecott, K.W. A human 5’-tyrosyl DNA phosphodiesterase that repairs topoisomerase-mediated DNA damage. Nature 2009, 461, 674–678. [Google Scholar] [CrossRef]
- Jones, S.A.; Boregowda, R.; Spratt, T.E.; Hu, J. In vitro epsilon RNA-dependent protein priming activity of human hepatitis B virus polymerase. J. Virol. 2012, 86, 5134–5150. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; McAllister, R.; Boregowda, R.; Sohn, J.A.; Cortes Ledesma, F.; Caldecott, K.W.; Seeger, C.; Hu, J. Does Tyrosyl DNA Phosphodiesterase-2 Play a Role in Hepatitis B Virus Genome Repair? PLoS ONE 2015, 10, e0128401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, B.Y.; Huang, T.S.; Pludwinski, E.; Heller, B.; Wojcik, F.; Lipkowitz, G.E.; Parekh, A.; Cho, C.; Shrirao, A.; Muir, T.W.; et al. Long-term hepatitis B infection in a scalable hepatic co-culture system. Nat. Commun. 2017, 8, 125. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, L.; Bambara, R.A. Flap endonuclease 1. Annu. Rev. Biochem. 2013, 82, 119–138. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Jia, J.; Finger, L.D.; Guo, Z.; Zer, C.; Shen, B. Functional regulation of FEN1 nuclease and its link to cancer. Nucleic Acids Res. 2011, 39, 781–794. [Google Scholar] [CrossRef] [Green Version]
- Gloor, J.W.; Balakrishnan, L.; Bambara, R.A. Flap endonuclease 1 mechanism analysis indicates flap base binding prior to threading. J. Biol. Chem. 2010, 285, 34922–34931. [Google Scholar] [CrossRef] [Green Version]
- Dorjsuren, D.; Kim, D.; Maloney, D.J.; Wilson, D.M., 3rd; Simeonov, A. Complementary non-radioactive assays for investigation of human flap endonuclease 1 activity. Nucleic Acids Res. 2011, 39, e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tumey, L.N.; Bom, D.; Huck, B.; Gleason, E.; Wang, J.; Silver, D.; Brunden, K.; Boozer, S.; Rundlett, S.; Sherf, B.; et al. The identification and optimization of a N-hydroxy urea series of flap endonuclease 1 inhibitors. Bioorg Med. Chem. Lett. 2005, 15, 277–281. [Google Scholar] [CrossRef]
- Newman, M.; Chua, P.K.; Tang, F.M.; Su, P.Y.; Shih, C. Testing an electrostatic interaction hypothesis of hepatitis B virus capsid stability by using an in vitro capsid disassembly/reassembly system. J. Virol. 2009, 83, 10616–10626. [Google Scholar] [CrossRef] [Green Version]
- Su, P.Y.; Yang, C.J.; Chu, T.H.; Chang, C.H.; Chiang, C.; Tang, F.M.; Lee, C.Y.; Shih, C. HBV maintains electrostatic homeostasis by modulating negative charges from phosphoserine and encapsidated nucleic acids. Sci Rep. 2016, 6, 38959. [Google Scholar] [CrossRef] [Green Version]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pourquier, P.; Jensen, A.D.; Gong, S.S.; Pommier, Y.; Rogler, C.E. Human DNA topoisomerase I-mediated cleavage and recombination of duck hepatitis B virus DNA in vitro. Nucleic Acids Res. 1999, 27, 1919–1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheraz, M.; Cheng, J.; Tang, L.; Chang, J.; Guo, J.T. Cellular DNA Topoisomerases Are Required for the Synthesis of Hepatitis B Virus Covalently Closed Circular DNA. J. Virol. 2019, 93, e02230-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murante, R.S.; Rumbaugh, J.A.; Barnes, C.J.; Norton, J.R.; Bambara, R.A. Calf RTH-1 nuclease can remove the initiator RNAs of Okazaki fragments by endonuclease activity. J. Biol. Chem. 1996, 271, 25888–25897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavis, J.E.; Lomonosova, E. The hepatitis B virus ribonuclease H as a drug target. Antivir. Res. 2015, 118, 132–138. [Google Scholar] [CrossRef] [Green Version]
- Hyjek, M.; Figiel, M.; Nowotny, M. RNases H: Structure and mechanism. DNA Repair 2019, 84, 102672. [Google Scholar] [CrossRef]
- Majka, J.; Burgers, P.M. The PCNA-RFC families of DNA clamps and clamp loaders. Prog. Nucleic Acid Res. Mol. Biol. 2004, 78, 227–260. [Google Scholar] [CrossRef]
- Yao, N.Y.; O’Donnell, M. The RFC clamp loader: Structure and function. Subcell BioChem. 2012, 62, 259–279. [Google Scholar] [CrossRef] [Green Version]
- Bruning, J.B.; Shamoo, Y. Structural and thermodynamic analysis of human PCNA with peptides derived from DNA polymerase-delta p66 subunit and flap endonuclease-1. Structure 2004, 12, 2209–2219. [Google Scholar] [CrossRef] [Green Version]
- Krishna, T.S.; Kong, X.P.; Gary, S.; Burgers, P.M.; Kuriyan, J. Crystal structure of the eukaryotic DNA polymerase processivity factor PCNA. Cell 1994, 79, 1233–1243. [Google Scholar] [CrossRef]
- Wright, G.E.; Hubscher, U.; Khan, N.N.; Focher, F.; Verri, A. Inhibitor analysis of calf thymus DNA polymerases alpha, delta and epsilon. FEBS Lett. 1994, 341, 128–130. [Google Scholar] [CrossRef] [Green Version]
- Sheaff, R.; Ilsley, D.; Kuchta, R. Mechanism of DNA polymerase alpha inhibition by aphidicolin. Biochemistry 1991, 30, 8590–8597. [Google Scholar] [CrossRef]
- Burgers, P.M.J.; Kunkel, T.A. Eukaryotic DNA Replication Fork. Annu. Rev. BioChem. 2017, 86, 417–438. [Google Scholar] [CrossRef]
- Kunkel, T.A.; Burgers, P.M.J. Arranging eukaryotic nuclear DNA polymerases for replication: Specific interactions with accessory proteins arrange Pols alpha, delta, and in the replisome for leading-strand and lagging-strand DNA replication. Bioessays 2017, 39. [Google Scholar] [CrossRef]
- Ogi, T.; Limsirichaikul, S.; Overmeer, R.M.; Volker, M.; Takenaka, K.; Cloney, R.; Nakazawa, Y.; Niimi, A.; Miki, Y.; Jaspers, N.G.; et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol. Cell 2010, 37, 714–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ellenberger, T.; Tomkinson, A.E. Eukaryotic DNA ligases: Structural and functional insights. Annu. Rev. BioChem. 2008, 77, 313–338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arakawa, H.; Bednar, T.; Wang, M.; Paul, K.; Mladenov, E.; Bencsik-Theilen, A.A.; Iliakis, G. Functional redundancy between DNA ligases I and III in DNA replication in vertebrate cells. Nucleic Acids Res. 2012, 40, 2599–2610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Chalony, C.; Hoffschir, F.; Gauthier, L.R.; Gross, J.; Biard, D.S.; Boussin, F.D.; Pennaneach, V. Partial complementation of a DNA ligase I deficiency by DNA ligase III and its impact on cell survival and telomere stability in mammalian cells. Cell Mol. Life Sci. 2012, 69, 2933–2949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Katyal, S.; Lee, Y.; Zhao, J.; Rehg, J.E.; Russell, H.R.; McKinnon, P.J. DNA ligase III is critical for mtDNA integrity but not Xrcc1-mediated nuclear DNA repair. Nature 2011, 471, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Simsek, D.; Furda, A.; Gao, Y.; Artus, J.; Brunet, E.; Hadjantonakis, A.K.; Van Houten, B.; Shuman, S.; McKinnon, P.J.; Jasin, M. Crucial role for DNA ligase III in mitochondria but not in Xrcc1-dependent repair. Nature 2011, 471, 245–248. [Google Scholar] [CrossRef]
- Guo, H.; Xu, C.; Zhou, T.; Block, T.M.; Guo, J.T. Characterization of the host factors required for hepadnavirus covalently closed circular (ccc) DNA formation. PLoS ONE 2012, 7, e43270. [Google Scholar] [CrossRef] [Green Version]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [Green Version]
- Polo, S.E.; Jackson, S.P. Dynamics of DNA damage response proteins at DNA breaks: A focus on protein modifications. Genes Dev. 2011, 25, 409–433. [Google Scholar] [CrossRef] [Green Version]
- Marechal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef]
- Blackford, A.N.; Jackson, S.P. ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [Green Version]
- Weitzman, M.D.; Fradet-Turcotte, A. Virus DNA Replication and the Host DNA Damage Response. Annu. Rev. Virol. 2018, 5, 141–164. [Google Scholar] [CrossRef]
- Lilley, C.E.; Schwartz, R.A.; Weitzman, M.D. Using or abusing: Viruses and the cellular DNA damage response. Trends MicroBiol. 2007, 15, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.H.; Hullinger, R.L.; Andrisani, O.M. Hepatitis B virus X protein via the p38MAPK pathway induces E2F1 release and ATR kinase activation mediating p53 apoptosis. J. Biol. Chem. 2008, 283, 25455–25467. [Google Scholar] [CrossRef] [Green Version]
- Ricardo-Lax, I.; Ramanan, V.; Michailidis, E.; Shamia, T.; Reuven, N.; Rice, C.M.; Shlomai, A.; Shaul, Y. Hepatitis B virus induces RNR-R2 expression via DNA damage response activation. J. Hepatol. 2015, 63, 789–796. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Luckenbaugh, L.; Hu, H.; Yan, Z.; Gao, L.; Hu, J. Involvement of Host ATR-CHK1 Pathway in Hepatitis B Virus Covalently Closed Circular DNA Formation. mBio 2020, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.Y.; Zhang, B.H.; Theele, D.; Litwin, S.; Toll, E.; Summers, J. Single-cell analysis of covalently closed circular DNA copy numbers in a hepadnavirus-infected liver. Proc. Natl. Acad. Sci. USA 2003, 100, 12372–12377. [Google Scholar] [CrossRef] [Green Version]
- Werle-Lapostolle, B.; Bowden, S.; Locarnini, S.; Wursthorn, K.; Petersen, J.; Lau, G.; Trepo, C.; Marcellin, P.; Goodman, Z.; Delaney, W.E., IV; et al. Persistence of cccDNA during the natural history of chronic hepatitis B and decline during adefovir dipivoxil therapy. Gastroenterology 2004, 126, 1750–1758. [Google Scholar] [CrossRef] [Green Version]
- Bourne, E.J.; Dienstag, J.L.; Lopez, V.A.; Sander, T.J.; Longlet, J.M.; Hall, J.G.; Kwiatkowski, R.W.; Wright, T.; Lai, C.L.; Condreay, L.D. Quantitative analysis of HBV cccDNA from clinical specimens: Correlation with clinical and virological response during antiviral therapy. J. Viral Hepat. 2007, 14, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Peng, B.; He, W.; Zhong, G.; Qi, Y.; Ren, B.; Gao, Z.; Jing, Z.; Song, M.; Xu, G.; et al. Molecular determinants of hepatitis B and D virus entry restriction in mouse sodium taurocholate cotransporting polypeptide. J. Virol. 2013, 87, 7977–7991. [Google Scholar] [CrossRef] [Green Version]
- Winer, B.Y.; Shirvani-Dastgerdi, E.; Bram, Y.; Sellau, J.; Low, B.E.; Johnson, H.; Huang, T.; Hrebikova, G.; Heller, B.; Sharon, Y.; et al. Preclinical assessment of antiviral combination therapy in a genetically humanized mouse model for hepatitis delta virus infection. Sci. Transl. Med. 2018, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lempp, F.A.; Mutz, P.; Lipps, C.; Wirth, D.; Bartenschlager, R.; Urban, S. Evidence that hepatitis B virus replication in mouse cells is limited by the lack of a host cell dependency factor. J. Hepatol. 2016, 64, 556–564. [Google Scholar] [CrossRef]
- Raney, A.K.; Eggers, C.M.; Kline, E.F.; Guidotti, L.G.; Pontoglio, M.; Yaniv, M.; McLachlan, A. Nuclear covalently closed circular viral genomic DNA in the liver of hepatocyte nuclear factor 1 alpha-null hepatitis B virus transgenic mice. J. Virol. 2001, 75, 2900–2911. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Guo, H.; Pan, X.B.; Mao, R.; Yu, W.; Xu, X.; Wei, L.; Chang, J.; Block, T.M.; Guo, J.T. Interferons accelerate decay of replication-competent nucleocapsids of hepatitis B virus. J. Virol. 2010, 84, 9332–9340. [Google Scholar] [CrossRef] [Green Version]
- Cui, X.; Guo, J.T.; Hu, J. Hepatitis B Virus Covalently Closed Circular DNA Formation in Immortalized Mouse Hepatocytes Associated with Nucleocapsid Destabilization. J. Virol. 2015, 89, 9021–9028. [Google Scholar] [CrossRef] [Green Version]
- Waga, S.; Hannon, G.J.; Beach, D.; Stillman, B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature 1994, 369, 574–578. [Google Scholar] [CrossRef] [PubMed]
- Marshall, A.; Rushbrook, S.; Davies, S.E.; Morris, L.S.; Scott, I.S.; Vowler, S.L.; Coleman, N.; Alexander, G. Relation between hepatocyte G1 arrest, impaired hepatic regeneration, and fibrosis in chronic hepatitis C virus infection. Gastroenterology 2005, 128, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.; Yadav, N.P.; Rai, V.K.; Sinha, P.; Yadav, K.S.; Jain, S.; Arora, S. Efficient hepatic delivery of drugs: Novel strategies and their significance. Biomed. Res. Int. 2013, 2013, 382184. [Google Scholar] [CrossRef] [Green Version]
- Erion, M.D.; van Poelje, P.D.; Mackenna, D.A.; Colby, T.J.; Montag, A.C.; Fujitaki, J.M.; Linemeyer, D.L.; Bullough, D.A. Liver-targeted drug delivery using HepDirect prodrugs. J. Pharmacol. Exp. Ther 2005, 312, 554–560. [Google Scholar] [CrossRef]
- Zhong, S.; Chen, X.; Zhu, X.; Dziegielewska, B.; Bachman, K.E.; Ellenberger, T.; Ballin, J.D.; Wilson, G.; Tomkinson, A.E.; MacKerell, A. Identification and validation of human DNA ligase inhibitors using computer-aided drug design. J. Med. Chem. 2008, 51, 4553–4562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Zhong, S.; Zhu, X.; Dziegielewska, B.; Ellenberger, T.; Wilson, G.; MacKerell, A.; Tomkinson, A.E. Rational design of human DNA ligase inhibitors that target cellular DNA replication and repair. Cancer Res. 2008, 68, 3169–3177. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Inhibitor | Target | Effects on cccDNA Biogenesis | Effective Dose Tested | System Used | References |
|---|---|---|---|---|---|
| Aphidicolin | DNA polymerases POLδ, POLα, and POLε | Specifically inhibits the synthesis of the plus strand | 100 μM | Biochemical | [19,75] |
| Reduced de novo cccDNA formation and amplification | 100–400 μM for de novo formation; 1 μM for intracellular amplification | hNTCP-HepG2 and HepAD38 cell lines | [33,75] | ||
| p21 peptide | PCNA-POLδ interaction | Specifically inhibits the synthesis of the plus strand | 100 μM | Biochemical | [19] |
| PTPD | FEN-1 endonuclease | Reduced de novo cccDNA formation and its amplification | 5–20 μM | hNTCP-HepG2 and Hep38.7-Tet cell lines | [19,77] |
| Topotecan | TOP1 | Reduced cccDNA intracellular amplification | 0.1–4 μM | HepAD38 | [104] |
| Camptothecin | TOP1 | Same as above | 0.06–2 μM | HepAD38 | [104] |
| Idarubicin | TOP2 | Same as above | 16–250 nM | HepAD38 | [104] |
| Doxorubincin | TOP2 | Same as above | 62–250 nM | HepAD38 | [104] |
| Aclarubicin | TOP2 | Same as above | 250–1000 nM | HepAD38 | [104] |
| Mitoxantrone | TOP2 | Same as above | 500 nM | HepAD38 | [104] |
| Merbarone | TOP2 | Same as above | 6–100 μM | HepAD38 | [104] |
| L1 | LIG1 and LIG3 | Inhibits cccDNA formation | 20 μM | Biochemical | [50,147,148] |
| L25 | LIG1 and LIG3 | Inhibits cccDNA formation | 25 μM | Biochemical | [50,147,148] |
| L189 | LIG1, LIG3, and LIG4 | Inhibits cccDNA formation | 50 μM | Biochemical | [50,147,148] |
| LIG1, LIG3, and LIG4 | Reduced cccDNA amplification in cell culture | 10–20 μM | Tet- inducible HepDG10 cells | [50,147,148] | |
| AZD6738 | ATR | Reduced de novo cccDNA formation and intracellular amplification | 25–50 μM | hNTCP-HepG2, AML12HBV10, and primary human hepatocytes | [133] |
| VE-821 | ATR | Reduced de novo cccDNA formation and intracellular amplification | 5–10 μM | hNTCP-HepG2, AML12HBV10 | [133] |
| CGK733 | ATM and ATR | Reduced de novo cccDNA formation | 1–12 μM | hNTCP-HepG2, and primary human hepatocytes | [133] |
| Torin2 | ATM and ATR | Reduced de novo cccDNA formation and intracellular amplification | 0.03–1 μM | hNTCP-HepG2, AML12HBV10, and primary human hepatocytes | [133] |
| PF477736 | CHK1 and CHK2 | Reduced cccDNA intracellular amplification | 8 μM | AML12HBV10 | [133] |
| CHIR-124 | CHK1 | Reduced de novo cccDNA formation and intracellular amplification | 1–4 μM | hNTCP-HepG2, HepAD38, AML12HBV10, and primary human hepatocytes | [133] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, L.; Ploss, A. Mechanism of Hepatitis B Virus cccDNA Formation. Viruses 2021, 13, 1463. https://doi.org/10.3390/v13081463
Wei L, Ploss A. Mechanism of Hepatitis B Virus cccDNA Formation. Viruses. 2021; 13(8):1463. https://doi.org/10.3390/v13081463
Chicago/Turabian StyleWei, Lei, and Alexander Ploss. 2021. "Mechanism of Hepatitis B Virus cccDNA Formation" Viruses 13, no. 8: 1463. https://doi.org/10.3390/v13081463
APA StyleWei, L., & Ploss, A. (2021). Mechanism of Hepatitis B Virus cccDNA Formation. Viruses, 13(8), 1463. https://doi.org/10.3390/v13081463