
Exploring the Cause of Diarrhoea and Poor Growth in 8–11-Week-Old Pigs from an Australian Pig Herd Using Metagenomic Sequencing


Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical History and Sample Collection
2.2. Enrichment, Nucleic Acid Extraction, cDNA Synthesis and Non-Targeted Amplification
2.3. Library Preparation, Next Generation Sequencing (NGS), and Detection of Different Virus Sequences
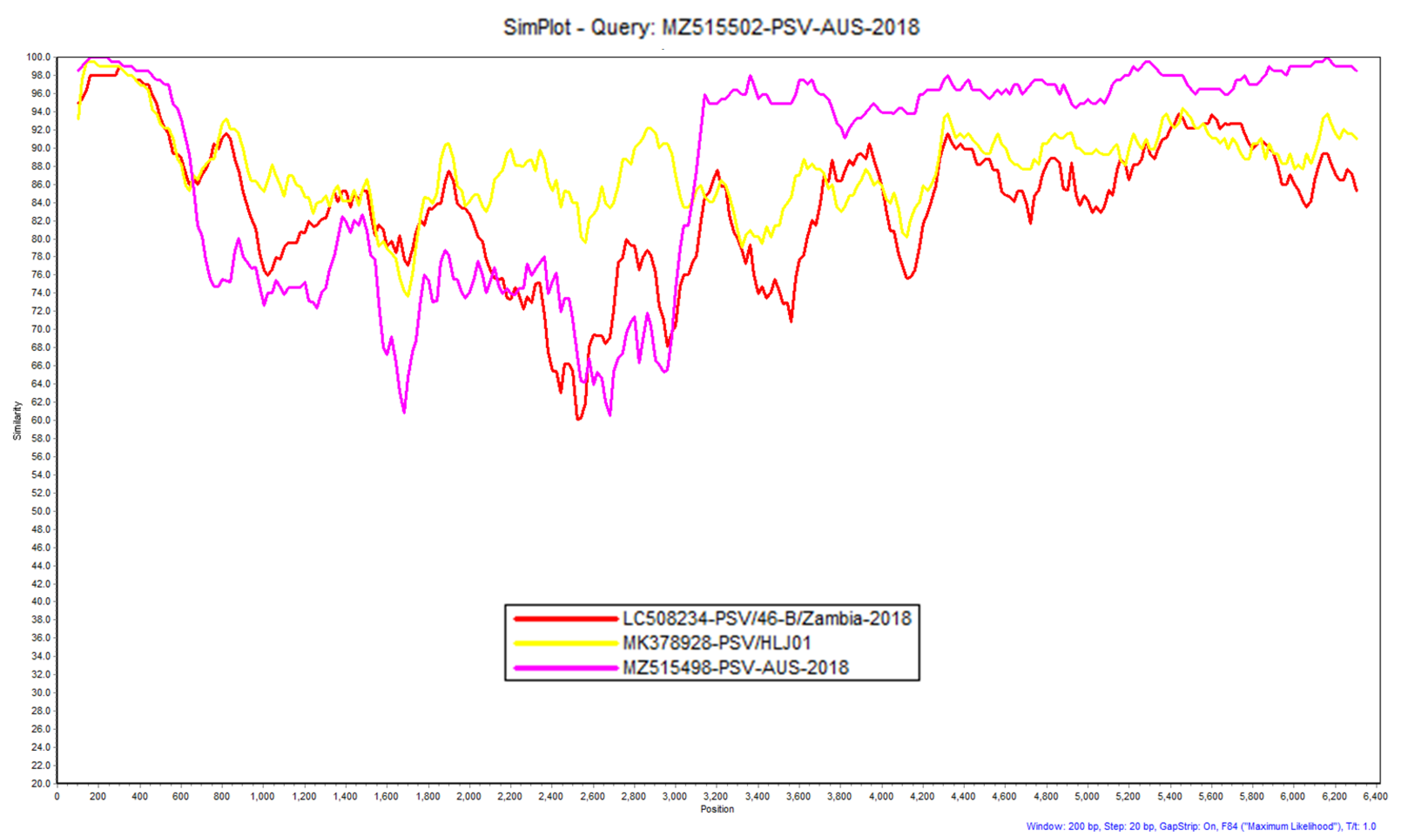
2.4. Similarity Plot (SimPlot) Analysis of Identified Virus Sequences
2.5. Mapping of Bacterial and Antimicrobial Resistance (AMR) Gene Reads
3. Results
3.1. Porcine Sapelovirus (PSV)
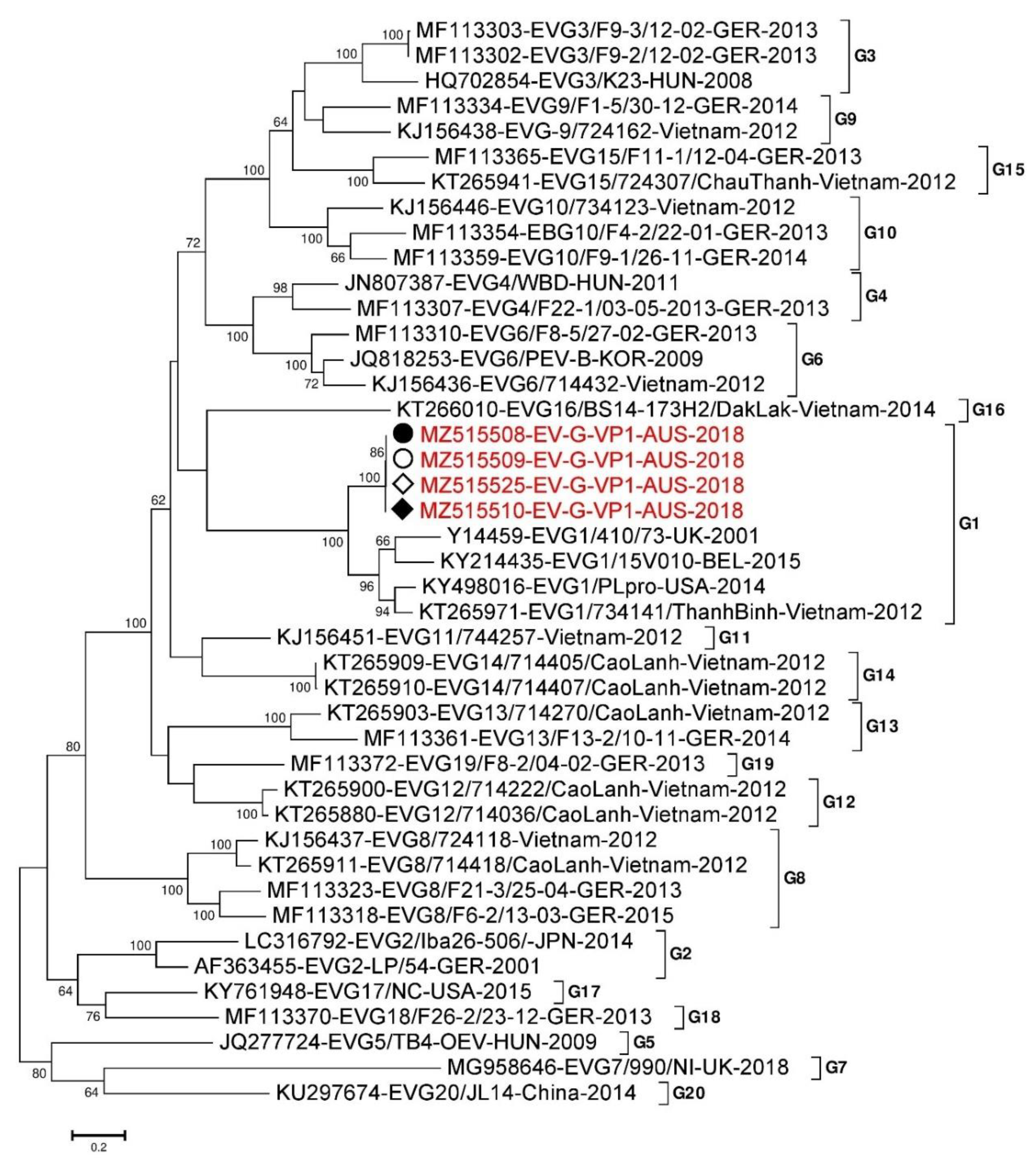
3.2. Porcine Enterovirus G (PEV-G)
3.3. Porcine Teschovirus (PTV)
3.4. Porcine Astrovirus (PAstV)
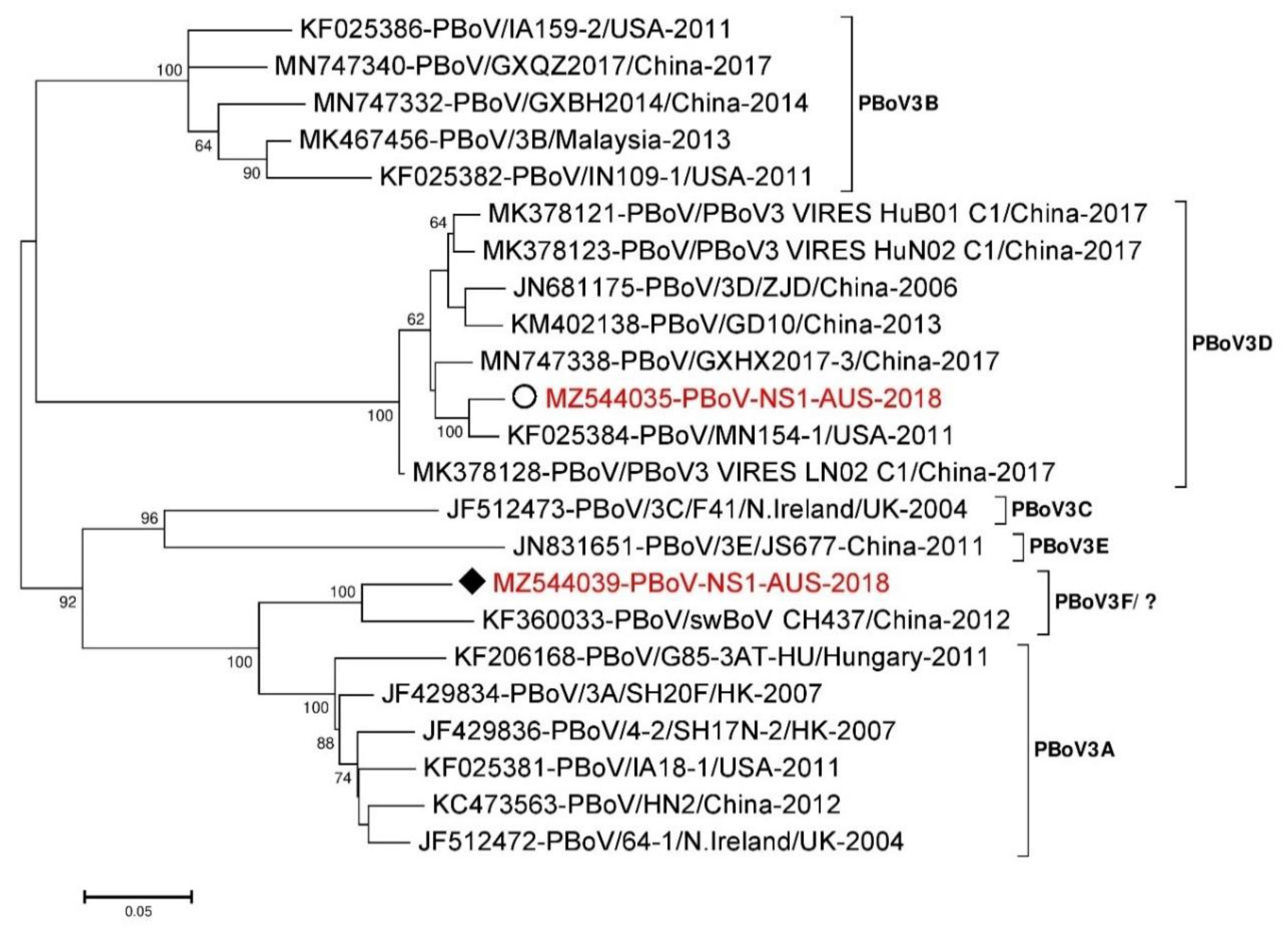
3.5. Porcine Bocavirus (PBoV)
3.6. Porcine Parvovirus 2 (PPV2)
3.7. Porcine Circovirus Type 2 (PCV2)
3.8. Porcine Torque Teno Sus Virus (TTSuV)
3.9. Analysis of Reads Mapping to Bacteria and to Antimicrobial Resistance (AMR) Genes
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arnold, M.; Crienen, A.; Swam, H.; Berg, S.V.; Jolie, R.; Nathues, H. Correlation of Lawsonia intracellularis positivity in quantitative PCR and herd factors in European pig herds. Porc. Health Manag. 2021, 7, 1–8. [Google Scholar] [CrossRef]
- Cornelison, A.; Karriker, L.; Williams, N.; Haberl, B.; Stalder, K.; Schulz, L.; Patience, J. Impact of health challenges on pig growth performance, carcass characteristics, and net returns under commercial conditions. Transl. Anim. Sci. 2018, 2, 50–61. [Google Scholar] [CrossRef] [Green Version]
- Sjolund, M.; Zoric, M.; Wallgren, P. Financial impact on pig production III: Gastrointestinal disorders. In Proceedings of the 6th European Symposium of Porcine Health Management, Sorrento, Italy, 7–9 May 2014. [Google Scholar]
- Dufresne, L. Economics of Pig Health Improvements. Available online: https://porkgateway.org/wp-content/uploads/2015/07/economics-of-pig-health-improvement1.pdf (accessed on 10 June 2021).
- Van Breda, L.K.; Dhungyel, O.P.; Ginn, A.N.; Iredell, J.R.; Ward, M.P. Pre-and post-weaning scours in southeastern Australia: A survey of 22 commercial pig herds and characterisation of Escherichia coli isolates. PLoS ONE 2017, 12, e0172528. [Google Scholar] [CrossRef] [Green Version]
- Schulz, L.L.; Tonsor, G.T. Assessment of the economic impacts of porcine epidemic diarrhoea virus in the United States. J. Anim. Sci. 2015, 93, 5111–5118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, R.; Almond, G.; Byers, E. Swine Diseases and Disorders. Encycl. Agric. Food Syst. 2014, 261–276. [Google Scholar] [CrossRef]
- VanderWaal, K.; Deen, J. Global trends in infectious diseases of swine. Proc. Natl. Acad. Sci. USA 2018, 115, 11495–11500. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, A. Diseases affecting pigs: An overview of common bacterial, viral and parasitic pathogens of pigs. In Achieving Sustainable Production of Pig Meat: Animal Health and Welfare; Alejandro, R., Ed.; Iowa State University: Ames, IA, USA, 2018; Volume 3, pp. 3–29. [Google Scholar]
- Bergeland, M.E.; Henry, S.C. Infectious diarrhoeas of young pigs. Vet. Clin. N. Am. Large Anim. Pract. 1982, 4, 389–399. [Google Scholar]
- Matias Ferreyra, F.; Arruda, B.; Stevenson, G.; Schwartz, K.; Madson, D.; Yoon, K.-J.; Zhang, J.; Piñeyro, P.; Chen, Q.; Arruda, P. Development of polioencephalomyelitis in cesarean-derived colostrum-deprived pigs following experimental inoculation with either teschovirus a serotype 2 or serotype 11. Viruses 2017, 9, 179. [Google Scholar] [CrossRef] [Green Version]
- Arruda, P.; Arruda, B.; Schwartz, K.; Vannucci, F.; Resende, T.; Rovira, A.; Sundberg, P.; Nietfeld, J.; Hause, B. Detection of a novel sapelovirus in central nervous tissue of pigs with polioencephalomyelitis in the USA. Transbound. Emerg. Dis. 2017, 64, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Wang, Y.; Shen, Q.; Zhang, W.; Hua, X. Prevalence of porcine enterovirus 9 in pigs in middle and eastern China. Virol. J. 2013, 10, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.-S.; Kang, M.-I.; Son, K.-Y.; Bak, G.-Y.; Park, J.-G.; Hosmillo, M.; Seo, J.-Y.; Kim, J.-Y.; Alfajaro, M.M.; Soliman, M. Pathogenesis of Korean Sapelovirus A in piglets and chicks. J. Gen. Virol. 2016, 97, 2566–2574. [Google Scholar] [CrossRef] [PubMed]
- Schock, A.; Gurrala, R.; Fuller, H.; Foyle, L.; Dauber, M.; Martelli, F.; Scholes, S.; Roberts, L.; Steinbach, F.; Dastjerdi, A. Investigation into an outbreak of encephalomyelitis caused by a neuroinvasive porcine sapelovirus in the United Kingdom. Vet. Microbiol. 2014, 172, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Padmanabhan, A.; Hause, B.M. Detection and characterization of a novel genotype of porcine astrovirus 4 from nasal swabs from pigs with acute respiratory disease. Arch. Virol. 2016, 161, 2575–2579. [Google Scholar] [CrossRef]
- Boros, Á.; Albert, M.; Pankovics, P.; Bíró, H.; Pesavento, P.A.; Phan, T.G.; Delwart, E.; Reuter, G. Outbreaks of neuroinvasive astrovirus associated with encephalomyelitis, weakness, and paralysis among weaned pigs, Hungary. Emerg. Infect. Dis. 2017, 23, 1982–1993. [Google Scholar] [CrossRef]
- Rawal, G.; Ferreyra, F.M.; Macedo, N.R.; Bradner, L.K.; Harmon, K.M.; Allison, G.; Linhares, D.C.; Arruda, B.L. Ecology of Porcine Astrovirus Type 3 in a Herd with Associated Neurologic Disease. Viruses 2020, 12, 992. [Google Scholar] [CrossRef]
- Batista, L. Porcine Circovirus Associated Diseases (PCVAD) in Canada-Prevalence, Co-Factors, and Risk Factors. In Advances in Pork Production, Proceedings of the 2007 Banff Pork Seminar, Banff, Alberta, Canada, 16–19 January 2007; University of Alberta: Edmonton, AB, Canada, 2007; Volume 18, pp. 57–68. [Google Scholar]
- Segalés, J. Porcine circovirus type 2 (PCV2) infections: Clinical signs, pathology and laboratory diagnosis. Virus Res. 2012, 164, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Phaneuf, L.R.; Ceccarelli, A.; Laing, J.R.; Moloo, B.; Turner, P.V. Porcine dermatitis and nephropathy syndrome associated with porcine circovirus 2 infection in a Yorkshire pig. J. Am. Assoc. Lab. Anim. Sci. 2007, 46, 68–72. [Google Scholar] [PubMed]
- Allan, G.M.; Ellis, J.A. Porcine circoviruses: A review. J. Vet. Diagn. Investig. 2000, 12, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Ruan, W.; Yue, H.; Tang, C.; Zhou, K.; Zhang, B. Viral communities associated with porcine respiratory disease complex in intensive commercial farms in Sichuan province, China. Sci. Rep. 2018, 8, 13341. [Google Scholar] [CrossRef]
- McMenamy, M.J.; McKillen, J.; McNair, I.; Duffy, C.; Blomström, A.-L.; Charreyre, C.; Welsh, M.; Allan, G. Detection of a porcine boca-like virus in combination with porcine circovirus type 2 genotypes and Torque teno sus virus in pigs from postweaning multisystemic wasting syndrome (PMWS)-affected and non-PMWS-affected farms in archival samples from Great Britain. Vet. Microbiol. 2013, 164, 293–298. [Google Scholar] [PubMed]
- McOrist, S.; Smith, S.; Green, L. Estimate of direct financial losses due to porcine proliferative enteropathy. Vet. Rec. 1997, 140, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Karuppannan, A.K.; Opriessnig, T. Lawsonia intracellularis: Revisiting the disease ecology and control of this fastidious pathogen in pigs. Front. Vet. Sci. 2018, 5, 181. [Google Scholar] [CrossRef]
- Vannucci, F.; Gebhart, C. Recent advances in understanding the pathogenesis of Lawsonia intracellularis infections. Vet. Pathol. 2014, 51, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Osorio, J.; Carvajal, A.; Naharro, G.; Rubio, P.; La, T.; Phillips, N.; Hampson, D. Identification of weakly haemolytic Brachyspira isolates recovered from pigs with diarrhoea in Spain and Portugal and comparison with results from other countries. Res. Vet. Sci. 2013, 95, 861–869. [Google Scholar] [CrossRef] [Green Version]
- Clothier, K.A.; Kinyon, J.M.; Frana, T.S.; Naberhaus, N.; Bower, L.; Strait, E.L.; Schwartz, K. Species characterization and minimum inhibitory concentration patterns of Brachyspira species isolates from swine with clinical disease. J. Vet. Diagn. Investig. 2011, 23, 1140–1145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampson, D.J.; La, T.; Phillips, N.D. Emergence of Brachyspira species and strains: Reinforcing the need for surveillance. Porc. Health Manag. 2015, 1, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Burrough, E.; Terhorst, S.; Sahin, O.; Zhang, Q. Prevalence of Campylobacter spp. relative to other enteric pathogens in grow-finish pigs with diarrhoea. Anaerobe 2013, 22, 111–114. [Google Scholar] [CrossRef]
- Edfors-Lilja, I.; Wallgren, P.; Axford, R.; Bishop, S.; Nicholas, F.; Owen, J. Escherichia coli and Salmonella diarrhoea in pigs. In Breedings for Disease Resistance in Farm Animals, 2nd ed.; CABI Publisher: Wallingford, UK, 2000; Volume 12, pp. 253–267. [Google Scholar]
- Suh, D.K.; Song, J.C. Simultaneous detection of Lawsonia intracellularis, Brachyspira hyodysenteriae and Salmonella spp. in swine intestinal specimens by multiplex polymerase chain reaction. J. Vet. Sci. 2005, 6, 231–237. [Google Scholar] [CrossRef]
- Leblanc-Maridor, M.; Beaudeau, F.; Seegers, H.; Denis, M.; Belloc, C. Rapid identification and quantification of Campylobacter coli and Campylobacter jejuni by real-time PCR in pure cultures and in complex samples. BMC Microbiol. 2011, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhatta, T.R.; Ryt-Hansen, P.; Nielsen, J.P.; Larsen, L.E.; Larsen, I.; Chamings, A.; Goecke, N.B.; Alexandersen, S. Infection Dynamics of Swine Influenza Virus in a Danish Pig Herd Reveals Recurrent Infections with Different Variants of the H1N2 Swine Influenza A Virus Subtype. Viruses 2020, 12, 1013. [Google Scholar] [CrossRef] [PubMed]
- Chelli, E.; De Sabato, L.; Vaccari, G.; Ostanello, F.; Di Bartolo, I. Detection and Characterization of Porcine Sapelovirus in Italian Pig Farms. Animals 2020, 10, 966. [Google Scholar] [CrossRef] [PubMed]
- Palmquist, J.M.; Munir, S.; Taku, A.; Kapur, V.; Goyal, S.M. Detection of porcine teschovirus and enterovirus type II by reverse transcription–polymerase chain reaction. J. Vet. Diagn. Investig. 2002, 14, 476–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Tang, W.; Hua, X. Molecular characterization of a porcine teschovirus HuN-1 isolate proliferating in PK-15 cell. BMC Vet. Res. 2018, 14, 1–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mor, S.K.; Chander, Y.; Marthaler, D.; Patnayak, D.P.; Goyal, S.M. Detection and molecular characterization of Porcine astrovirus strains associated with swine diarrhoea. J. Vet. Diagn. Investig. 2012, 24, 1064–1067. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Li, Y.; Sun, D.; Xia, Y.; Huang, J.; Guo, L. Detection and genetic analysis of porcine bocavirus in different swine herds in North Central China. Sci. World J. 2014, 2014. [Google Scholar] [CrossRef]
- Jacob, D.M.; Lee, C.Y.; Arshad, S.S.; Selvarajah, G.T.; Bande, F.; Ong, B.L.; Ooi, P.T. First molecular detection of porcine bocavirus in Malaysia. Trop. Anim. Health Prod. 2018, 50, 733–739. [Google Scholar] [CrossRef]
- Yang, K.; Jiao, Z.; Zhou, D.; Guo, R.; Duan, Z.; Tian, Y. Development of a multiplex PCR to detect and discriminate porcine circoviruses in clinical specimens. BMC Infect. Dis. 2019, 19, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Blomström, A.-L. Viral metagenomics as an emerging and powerful tool in veterinary medicine. Vet. Q. 2011, 31, 107–114. [Google Scholar] [CrossRef]
- Bhatta, T.R.; Chamings, A.; Vibin, J.; Alexandersen, S. Detection and characterisation of canine astrovirus, canine parvovirus and canine papillomavirus in puppies using next generation sequencing. Sci. Rep. 2019, 9, 1–10. [Google Scholar]
- Vibin, J.; Chamings, A.; Collier, F.; Klaassen, M.; Nelson, T.M.; Alexandersen, S. Metagenomics detection and characterisation of viruses in faecal samples from Australian wild birds. Sci. Rep. 2018, 8, 1–23. [Google Scholar] [CrossRef]
- Vibin, J.; Chamings, A.; Klaassen, M.; Bhatta, T.R.; Alexandersen, S. Metagenomic characterisation of avian parvoviruses and picornaviruses from Australian wild ducks. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Blomström, A.-L.; Fossum, C.; Wallgren, P.; Berg, M. Viral metagenomic analysis displays the co-infection situation in healthy and PMWS affected pigs. PLoS ONE 2016, 11, e0166863. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Kuroda, M.; Masuda, T.; Akagami, M.; Haga, K.; Tsuchiaka, S.; Kishimoto, M.; Naoi, Y.; Sano, K.; Omatsu, T. Whole genome analysis of porcine astroviruses detected in Japanese pigs reveals genetic diversity and possible intra-genotypic recombination. Infect. Genet. Evol. 2017, 50, 38–48. [Google Scholar] [CrossRef]
- Tsuchiaka, S.; Naoi, Y.; Imai, R.; Masuda, T.; Ito, M.; Akagami, M.; Ouchi, Y.; Ishii, K.; Sakaguchi, S.; Omatsu, T. Genetic diversity and recombination of enterovirus G strains in Japanese pigs: High prevalence of strains carrying a papain-like cysteine protease sequence in the enterovirus G population. PLoS ONE 2018, 13, e0190819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harima, H.; Kajihara, M.; Simulundu, E.; Bwalya, E.; Qiu, Y.; Isono, M.; Okuya, K.; Gonzalez, G.; Yamagishi, J.; Hang’ombe, B.M. Genetic and biological diversity of porcine Sapeloviruses prevailing in Zambia. Viruses 2020, 12, 180. [Google Scholar] [CrossRef] [Green Version]
- Wu, Z.; Ren, X.; Yang, L.; Hu, Y.; Yang, J.; He, G.; Zhang, J.; Dong, J.; Sun, L.; Du, J. Virome analysis for identification of novel mammalian viruses in bat species from Chinese provinces. J. Virol. 2012, 86, 10999–11012. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Pesavento, P.A.; Shan, T.; Leutenegger, C.M.; Wang, C.; Delwart, E. Viruses in diarrhoeic dogs include novel kobuviruses and sapoviruses. J. Gen. Virol. 2011, 92, 2534–2541. [Google Scholar] [CrossRef]
- Moreno, P.S.; Wagner, J.; Mansfield, C.S.; Stevens, M.; Gilkerson, J.R.; Kirkwood, C.D. Characterisation of the canine faecal virome in healthy dogs and dogs with acute diarrhoea using shotgun metagenomics. PLoS ONE 2017, 12, e0178433. [Google Scholar] [CrossRef] [Green Version]
- Phan, T.G.; Kapusinszky, B.; Wang, C.; Rose, R.K.; Lipton, H.L.; Delwart, E.L. The fecal viral flora of wild rodents. PLoS Pathog. 2011, 7, e1002218. [Google Scholar] [CrossRef] [Green Version]
- Victoria, J.G.; Kapoor, A.; Li, L.; Blinkova, O.; Slikas, B.; Wang, C.; Naeem, A.; Zaidi, S.; Delwart, E. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J. Virol. 2009, 83, 4642–4651. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Yang, F.; Ren, L.; Xiong, Z.; Wu, Z.; Dong, J.; Sun, L.; Zhang, T.; Hu, Y.; Du, J. Unbiased parallel detection of viral pathogens in clinical samples by use of a metagenomic approach. J. Clin. Microbiol. 2011, 49, 3463–3469. [Google Scholar] [CrossRef] [Green Version]
- Bhatta, T.R.; Chamings, A.; Vibin, J.; Klaassen, M.; Alexandersen, S. Detection of a reassortant H9N2 avian influenza virus with intercontinental gene segments in a resident australian chestnut teal. Viruses 2020, 12, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mount, D.W. Using the basic local alignment search tool (BLAST). Cold Spring Harb. Protoc. 2007, 2007, 17. [Google Scholar] [CrossRef] [PubMed]
- Caboche, S.; Audebert, C.; Lemoine, Y.; Hot, D. Comparison of mapping algorithms used in high-throughput sequencing: Application to Ion Torrent data. BMC Genom. 2014, 15, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative Genomics Viewer (IGV): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Martin, D.; Posada, D.; Crandall, K.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar] [CrossRef] [Green Version]
- Conserved Protein Domain Family DUF3724. Available online: https://www.ncbi.nlm.nih.gov/Structure/cdd/cddsrv.cgi?uid=152955 (accessed on 20 July 2021).
- Van Dung, N.; Anh, P.H.; Van Cuong, N.; Hoa, N.T.; Carrique-Mas, J.; Hien, V.B.; Campbell, J.; Baker, S.; Farrar, J.; Woolhouse, M.E. Prevalence, genetic diversity and recombination of species G enteroviruses infecting pigs in Vietnam. J. Gen. Virol. 2014, 95, 549–556. [Google Scholar] [CrossRef]
- Vilar, M.; Peralta, B.; García-Bocanegra, I.; Simon-Grifé, M.; Bensaid, A.; Casal, J.; Segalés, J.; Pina-Pedrero, S. Distribution and genetic characterization of Enterovirus G and Sapelovirus A in six Spanish swine herds. Virus Res. 2016, 215, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Bunke, J.; Receveur, K.; Oeser, A.C.; Fickenscher, H.; Zell, R.; Krumbholz, A. High genetic diversity of porcine enterovirus G in Schleswig-Holstein, Germany. Arch. Virol. 2018, 163, 489–493. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Usherwood, E.; Nash, A. Lymphocyte recognition of picornaviruses. J. Gen. Virol. 1995, 76, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Kaku, Y.; Murakami, Y.; Sarai, A.; Wang, Y.; Ohashi, S.; Sakamoto, K. Antigenic properties of porcine teschovirus 1 (PTV-1) Talfan strain and molecular strategy for serotyping of PTVs. Arch. Virol. 2007, 152, 929–940. [Google Scholar] [CrossRef]
- Cano-Gómez, C.; Palero, F.; Buitrago, M.D.; García-Casado, M.A.; Fernández-Pinero, J.; Fernández-Pacheco, P.; Agüero, M.; Gómez-Tejedor, C.; Jiménez-Clavero, M.Á. Analyzing the genetic diversity of teschoviruses in Spanish pig populations using complete VP1 sequences. Infect. Genet. Evol. 2011, 11, 2144–2150. [Google Scholar] [CrossRef]
- De Benedictis, P.; Schultz-Cherry, S.; Burnham, A.; Cattoli, G. Astrovirus infections in humans and animals–molecular biology, genetic diversity, and interspecies transmissions. Infect. Genet. Evol. 2011, 11, 1529–1544. [Google Scholar] [CrossRef]
- Dong, J.; Dong, L.; Méndez, E.; Tao, Y. Crystal structure of the human astrovirus capsid spike. Proc. Natl. Acad. Sci. USA 2011, 108, 12681–12686. [Google Scholar] [CrossRef] [Green Version]
- Matsui, M.; Ushijima, H.; Hachiya, M.; Kakizawa, J.; Wen, L.; Oseto, M.; Morooka, K.; Kurtz, J.B. Determination of serotypes of astroviruses by reverse transcription-polymerase chain reaction and homologies of the types by the sequencing of Japanese isolates. Microbiol. Immunol. 1998, 42, 539–547. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.; Woo, P.C.; Yip, C.C.; Li, K.S.; Fu, C.T.; Huang, Y.; Chan, K.-H.; Yuen, K.-Y. Co-existence of multiple strains of two novel porcine bocaviruses in the same pig, a previously undescribed phenomenon in members of the family Parvoviridae, and evidence for inter-and intra-host genetic diversity and recombination. J. Gen. Virol. 2011, 92, 2047–2059. [Google Scholar] [CrossRef] [PubMed]
- McKillen, J.; McNeilly, F.; Duffy, C.; McMenamy, M.; McNair, I.; Hjertner, B.; Millar, A.; McKay, K.; Lagan, P.; Adair, B. Isolation in cell cultures and initial characterisation of two novel bocavirus species from swine in Northern Ireland. Vet. Microbiol. 2011, 152, 39–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.; Ma, J.; Xiao, S.; Fang, L.; Zeng, S.; Wen, L.; Zhang, X.; Ni, Y.; Guo, R.; Yu, Z. Complete genome sequence of a novel species of porcine bocavirus, PBoV5. Am. Soc. Microbiol. 2012. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.-H.; Xiao, C.-T.; Yin, S.-H.; Gerber, P.F.; Halbur, P.G.; Opriessnig, T. High prevalence and genetic diversity of porcine bocaviruses in pigs in the USA, and identification of multiple novel porcine bocaviruses. J. Gen. Virol. 2014, 95, 453–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, C.-T.; Halbur, P.G.; Opriessnig, T. Molecular evolutionary genetic analysis of emerging parvoviruses identified in pigs. Infect. Genet. Evol. 2013, 16, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.-Z.; Yu, J.-M.; Li, J.-S.; Cheng, W.-X.; Huang, C.-P.; Duan, Z.-J. Genome characterization of a novel porcine bocavirus. Arch. Virol. 2012, 157, 2125–2132. [Google Scholar] [CrossRef]
- Wang, E.; Liu, W.; Yang, B.; Liu, J.; Ma, X.; Lan, X. Complete sequence and phylogenetic analysis of a porcine bocavirus strain swBoV CH437. Virus Genes 2014, 48, 387–390. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T.-A. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Yang, N.; Li, J.; Yang, Q.; Qiao, J.; Cui, D.; Liu, F.; Li, H.; Zhou, S. Reduced antigen presentation capability and modified inflammatory/immunosuppressive cytokine expression of induced monocyte-derived dendritic cells from peripheral blood of piglets infected with porcine circovirus type 2. Arch. Virol. 2018, 163, 1231–1239. [Google Scholar] [CrossRef]
- Olvera, A.; Cortey, M.; Segales, J. Molecular evolution of porcine circovirus type 2 genomes: Phylogeny and clonality. Virology 2007, 357, 175–185. [Google Scholar] [CrossRef] [Green Version]
- Ramos, N.; Mirazo, S.; Castro, G.; Arbiza, J. Molecular analysis of Porcine Circovirus Type 2 strains from Uruguay: Evidence for natural occurring recombination. Infect. Genet. Evol. 2013, 19, 23–31. [Google Scholar] [CrossRef]
- Cortey, M.; Pileri, E.; Segalés, J.; Kekarainen, T. Globalisation and global trade influence molecular viral population genetics of Torque Teno Sus Viruses 1 and 2 in pigs. Vet. Microbiol. 2012, 156, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Sunaga, F.; Masuda, T.; Ito, M.; Akagami, M.; Naoi, Y.; Sano, K.; Katayama, Y.; Omatsu, T.; Oba, M.; Sakaguchi, S. Complete genomic analysis and molecular characterization of Japanese porcine sapeloviruses. Virus Genes 2019, 55, 198–208. [Google Scholar] [CrossRef] [PubMed]
- Prodělalová, J. The survey of porcine teschoviruses, sapeloviruses and enteroviruses B infecting domestic pigs and wild boars in the Czech Republic between 2005 and 2011. Infect. Genet. Evol. 2012, 12, 1447–1451. [Google Scholar] [CrossRef] [PubMed]
- Boros, Á.; Pankovics, P.; Reuter, G. Characterization of a novel porcine enterovirus in domestic pig in Hungary. Infect. Genet. Evol. 2011, 11, 1096–1102. [Google Scholar] [CrossRef]
- Zhang, W.; Yang, S.; Shen, Q.; Ren, L.; Shan, T.; Wei, J.; Cui, L.; Hua, X. Complete genome sequence of a novel porcine enterovirus strain in China. J. Virol. 2012, 86, 7008–7009. [Google Scholar] [CrossRef] [Green Version]
- Moon, H.-J.; Song, D.; Seon, B.H.; Kim, H.-K.; Park, S.-J.; An, D.-J.; Kim, J.-M.; Kang, B.-K.; Park, B.-K. Complete genome analysis of porcine enterovirus B isolated in Korea. J. Virol. 2012, 86, 10250. [Google Scholar] [CrossRef] [Green Version]
- Knutson, T.P.; Velayudhan, B.T.; Marthaler, D.G. A porcine enterovirus G associated with enteric disease contains a novel papain-like cysteine protease. J. Gen. Virol. 2017, 98, 1305–1310. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, C. First detection of novel enterovirus G recombining a torovirus papain–like protease gene associated with diarrhoea in swine in South Korea. Transbound. Emerg. Dis. 2019, 66, 1023–1028. [Google Scholar] [CrossRef] [Green Version]
- Malik, Y.S.; Bhat, S.; Vlasova, A.N.; Wang, F.-I.; Touil, N.; Ghosh, S.; Dhama, K.; Yadav, M.P.; Singh, R.K. Emerging and Transboundary Animal Viruses; Springer: Singapore, 2020; pp. 123–136. [Google Scholar]
- Forman, A.; Pass, D.; Connaughton, I. The characterisation and pathogenicity of porcine enteroviruses isolated in Victoria. Aust. Vet. J. 1982, 58, 136–142. [Google Scholar] [CrossRef]
- Xiao, C.-T.; Gimenez-Lirola, L.G.; Gerber, P.F.; Jiang, Y.-H.; Halbur, P.G.; Opriessnig, T. Identification and characterization of novel porcine astroviruses (PAstVs) with high prevalence and frequent co-infection of individual pigs with multiple PAstV types. J. Gen. Virol. 2013, 94, 570–582. [Google Scholar] [CrossRef]
- Lv, S.-L.; Zhang, H.-H.; Li, J.-Y.; Hu, W.-Q.; Song, Y.-T.; Opriessnig, T.; Xiao, C.-T. High genetic diversity and recombination events of porcine astrovirus strains identified from ill and asymptomatic pigs in 2017, Hunan Province, China. Virus Genes 2019, 55, 673–681. [Google Scholar] [CrossRef]
- Fang, Q.; Wang, C.; Liu, H.; Wu, Q.; Liang, S.; Cen, M.; Dong, Q.; Wei, Y.; Chen, Y.; Ouyang, K. Pathogenic characteristics of a porcine astrovirus strain isolated in China. Viruses 2019, 11, 1156. [Google Scholar] [CrossRef] [Green Version]
- Austermann-Busch, S.; Becher, P. RNA structural elements determine frequency and sites of nonhomologous recombination in an animal plus-strand RNA virus. J. Virol. 2012, 86, 7393–7402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simmonds, P. Recombination and selection in the evolution of picornaviruses and other mammalian positive-stranded RNA viruses. J. Virol. 2006, 80, 11124–11140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wohlgemuth, N.; Honce, R.; Schultz-Cherry, S. Astrovirus evolution and emergence. Infect. Genet. Evol. 2019, 69, 30–37. [Google Scholar] [CrossRef]
- Van Dung, N.; Anh, P.H.; Van Cuong, N.; Hoa, N.T.; Carrique-Mas, J.; Hien, V.B.; Sharp, C.; Rabaa, M.; Berto, A.; Campbell, J. Large-scale screening and characterization of enteroviruses and kobuviruses infecting pigs in Vietnam. J. Gen. Virol. 2016, 97, 378–388. [Google Scholar] [CrossRef]
- Qiu, Z.; Wang, Z.; Zhang, B.; Zhang, J.; Cui, S. The prevalence of porcine teschovirus in the pig population in northeast of China. J. Virol. Methods 2013, 193, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Wang, D.; Fang, L.; Ma, J.; Song, T.; Zhang, R.; Chen, H.; Xiao, S. Complete coding sequences and phylogenetic analysis of porcine bocavirus. J. Gen. Virol. 2011, 92, 784–788. [Google Scholar] [CrossRef] [Green Version]
- Blomström, A.-L.; Belák, S.; Fossum, C.; Fuxler, L.; Wallgren, P.; Berg, M. Studies of porcine circovirus type 2, porcine boca-like virus and torque teno virus indicate the presence of multiple viral infections in postweaning multisystemic wasting syndrome pigs. Virus Res. 2010, 152, 59–64. [Google Scholar] [CrossRef]
- Brunborg, I.M.; Moldal, T.; Jonassen, C.M. Quantitation of porcine circovirus type 2 isolated from serum/plasma and tissue samples of healthy pigs and pigs with postweaning multisystemic wasting syndrome using a TaqMan-based real-time PCR. J. Virol. Methods 2004, 122, 171–178. [Google Scholar] [CrossRef]
- Monini, M.; Vignolo, E.; Ianiro, G.; Ostanello, F.; Ruggeri, F.M.; Di Bartolo, I. Detection of Torque teno sus virus in pork bile and liver sausages. Food Environ. Virol. 2016, 8, 283–288. [Google Scholar] [CrossRef] [PubMed]
- Pfankuche, V.M.; Bodewes, R.; Hahn, K.; Puff, C.; Beineke, A.; Habierski, A.; Osterhaus, A.D.; Baumgärtner, W. Porcine bocavirus infection associated with encephalomyelitis in a pig, Germany. Emerg. Infect. Dis. 2016, 22, 1310–1312. [Google Scholar] [CrossRef] [Green Version]
- Correa-Fiz, F.; Franzo, G.; Llorens, A.; Huerta, E.; Sibila, M.; Kekarainen, T.; Segalés, J. Porcine circovirus 2 (PCV2) population study in experimentally infected pigs developing PCV2-systemic disease or a subclinical infection. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, T.; Zhang, X.; Liu, X.; Ren, L. Co-infection of swine with porcine circovirus type 2 and other swine viruses. Viruses 2019, 11, 185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rammohan, L.; Xue, L.; Wang, C.; Chittick, W.; Ganesan, S.; Ramamoorthy, S. Increased prevalence of torque teno viruses in porcine respiratory disease complex affected pigs. Vet. Microbiol. 2012, 157, 61–68. [Google Scholar] [CrossRef]
- Kekarainen, T.; Segalés, J. Torque teno sus virus in pigs: An emerging pathogen? Transbound. Emerg. Dis. 2012, 59, 103–108. [Google Scholar] [CrossRef]
- Rosell, C.; Segalés, J.; Plana-Duran, J.; Balasch, M.; Rodrıguez-Arrioja, G.; Kennedy, S.; Allan, G.; McNeilly, F.; Latimer, K.; Domingo, M. Pathological, immunohistochemical, and in-situ hybridization studies of natural cases of postweaning multisystemic wasting syndrome (PMWS) in pigs. J. Comp. Pathol. 1999, 120, 59–78. [Google Scholar] [CrossRef]
- Kekarainen, T.; Segalés, J. Porcine circovirus 2 immunology and viral evolution. Porc. Health Manag. 2015, 1, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Segalés, J. Best practice and future challenges for vaccination against porcine circovirus type 2. Expert Rev. Vaccines 2015, 14, 473–487. [Google Scholar] [CrossRef]
- Fachinger, V.; Bischoff, R.; Jedidia, S.B.; Saalmüller, A.; Elbers, K. The effect of vaccination against porcine circovirus type 2 in pigs suffering from porcine respiratory disease complex. Vaccine 2008, 26, 1488–1499. [Google Scholar] [CrossRef]
- Ren, X.; Tao, Y.; Cui, J.; Suo, S.; Cong, Y.; Tijssen, P. Phylogeny and evolution of porcine parvovirus. Virus Res. 2013, 178, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Streck, A.F.; Canal, C.W.; Truyen, U. Molecular epidemiology and evolution of porcine parvoviruses. Infect. Genet. Evol. 2015, 36, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Treacy, D. A genetic analysis of the pedigree Landrace pig breed in Australia. Aust. J. Exp. Agric. 1976, 16, 76–81. [Google Scholar] [CrossRef]
- Pig Breeds in Australia. Available online: https://www.dpi.nsw.gov.au/__data/assets/pdf_file/0007/872683/Pig-breeds-brochure.pdf (accessed on 21 June 2021).
- Firth, C.; Charleston, M.A.; Duffy, S.; Shapiro, B.; Holmes, E.C. Insights into the evolutionary history of an emerging livestock pathogen: Porcine circovirus 2. J. Virol. 2009, 83, 12813–12821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franzo, G.; Cortey, M.; Segalés, J.; Hughes, J.; Drigo, M. Phylodynamic analysis of porcine circovirus type 2 reveals global waves of emerging genotypes and the circulation of recombinant forms. Mol. Phylogenetics Evol. 2016, 100, 269–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naccache, S.N.; Greninger, A.L.; Lee, D.; Coffey, L.L.; Phan, T.; Rein-Weston, A.; Aronsohn, A.; Hackett, J.; Delwart, E.L.; Chiu, C.Y. The perils of pathogen discovery: Origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J. Virol. 2013, 87, 11966–11977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Dea, M.; Kabay, M.; Carr, J.; Wilcox, G.; Richards, R. Porcine circovirus–associated disease in weaner pigs in Western Australia. Aust. Vet. J. 2011, 89, 122–130. [Google Scholar] [CrossRef]
- Verani, M.; Casini, B.; Battistini, R.; Pizzi, F.; Rovini, E.; Carducci, A. One-year monthly monitoring of Torque teno virus (TTV) in river water in Italy. Water Sci. Technol. 2006, 54, 191–195. [Google Scholar] [CrossRef]
- Takayama, S.; Miura, T.; Matsuo, S.; Taki, M.; Sugii, S. Prevalence and persistence of a novel DNA TT virus (TTV) infection in Japanese haemophiliacs. Br. J. Haematol. 1999, 104, 626–629. [Google Scholar] [CrossRef]
- López-Lorenzo, G.; Díaz-Cao, J.M.; Prieto, A.; López-Novo, C.; López, C.M.; Díaz, P.; Rodríguez-Vega, V.; Díez-Baños, P.; Fernández, G. Environmental distribution of Porcine Circovirus Type 2 (PCV2) in swine herds with natural infection. Sci. Rep. 2019, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Patterson, A.; Opriessnig, T. Epidemiology and horizontal transmission of porcine circovirus type 2 (PCV2). Anim. Health Res. Rev. 2010, 11, 217–234. [Google Scholar] [CrossRef]
- Scott, A.; McCluskey, B.; Brown-Reid, M.; Grear, D.; Pitcher, P.; Ramos, G.; Spencer, D.; Singrey, A. Porcine epidemic diarrhoea virus introduction into the United States: Root cause investigation. Prev. Vet. Med. 2016, 123, 192–201. [Google Scholar] [CrossRef] [Green Version]
- Dee, S.; Neill, C.; Singrey, A.; Clement, T.; Cochrane, R.; Jones, C.; Patterson, G.; Spronk, G.; Christopher-Hennings, J.; Nelson, E. Modeling the transboundary risk of feed ingredients contaminated with porcine epidemic diarrhoea virus. BMC Vet. Res. 2016, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Nagai, M.; Wang, Q.; Oka, T.; Saif, L.J. Porcine sapoviruses: Pathogenesis, epidemiology, genetic diversity, and diagnosis. Virus Res. 2020, 286, 198025. [Google Scholar] [CrossRef] [PubMed]
- Bak, G.-Y.; Kang, M.-I.; Son, K.-Y.; Park, J.-G.; Kim, D.-S.; Seo, J.-Y.; Kim, J.-Y.; Alfajaro, M.M.; Soliman, M.; Baek, Y.-B. Occurrence and molecular characterization of Sapelovirus A in diarrhoea and non-diarrhoea feces of different age group pigs in one Korean pig farm. J. Vet. Med. Sci. 2016, 78, 1911–1914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+ C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Abundance of Virus Reads (%) | Identity with Closest Nucleotide Sequence in NCBI at Structural Region | Identity with Closest Nucleotide Sequence in NCBI at Non-Structural Region | ||||
|---|---|---|---|---|---|---|
| Samples | ||||||
| Pig 45, Colon | Pig 45, Lung | Pig 46, Colon | Pig 46, Lung | |||
| PC45-BC24-AUS-2018 | PL45-BC23-AUS-2018 | PC46-BC26-AUS-2018 | PL46-BC25-AUS-2018 | |||
| Virus Name | ||||||
| Porcine sapelovirus (PSV) | 0.3565 | 0.1805 | 0.9866 | 0.0235 | ~80–88% | ~89% |
| Porcine enterovirus G (PEV-G) | 0.2250 | 0.3537 | 0.1056 | 0.0238 | ~80% | ~83–85% |
| Porcine teschovirus (PTV) | 0.0160 | - | 0.0244 | 0.0072 | ~82–86% | ~87–89% |
| Porcine astrovirus (PAstV) | - | - | 0.0180 | - | ~75–81% | ~95% |
| Porcine bocavirus (PBoV) | 0.0051 | 0.0305 | 0.2246 | 0.0018 | ~78–90% | ~93–97% |
| Porcine parvovirus 2 (PPV2) | - | 0.0279 | - | 0.0001 | ~94% | ~95% |
| Porcine parvovirus 7 (PPV7) | 0.0002 | - | 0.0038 | 0.0041 | ~98% | ~99% |
| Porcine bufa virus (PBuV) | 0.0068 | - | 0.0022 | 0.0007 | ~94–99% | ~99% |
| Adeno associated virus (AAV) | 0.0013 | 0.0057 | 0.0023 | 0.0002 | ~80–97% | ~84–88% |
| Porcine circovirus 2 (PCV2) | 0.0002 | 0.00007 | 0.0007 | 0.00003 | 99.82% | 100% |
| Torque teno sus virus k2a (TTSuVk2a) | 0.0024 | 0.0119 | - | 0.00047 | ~97% (overlapped) | |
| Torque teno sus virus k2b (TTSuVk2b) | 0.0058 | 0.0249 | - | 0.013 | 99.92% (overlapped) | |
| Total | 0.6193 | 0.6351 | 1.3682 | 0.0749 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatta, T.R.; Chamings, A.; Alexandersen, S. Exploring the Cause of Diarrhoea and Poor Growth in 8–11-Week-Old Pigs from an Australian Pig Herd Using Metagenomic Sequencing. Viruses 2021, 13, 1608. https://doi.org/10.3390/v13081608
Bhatta TR, Chamings A, Alexandersen S. Exploring the Cause of Diarrhoea and Poor Growth in 8–11-Week-Old Pigs from an Australian Pig Herd Using Metagenomic Sequencing. Viruses. 2021; 13(8):1608. https://doi.org/10.3390/v13081608
Chicago/Turabian StyleBhatta, Tarka Raj, Anthony Chamings, and Soren Alexandersen. 2021. "Exploring the Cause of Diarrhoea and Poor Growth in 8–11-Week-Old Pigs from an Australian Pig Herd Using Metagenomic Sequencing" Viruses 13, no. 8: 1608. https://doi.org/10.3390/v13081608
APA StyleBhatta, T. R., Chamings, A., & Alexandersen, S. (2021). Exploring the Cause of Diarrhoea and Poor Growth in 8–11-Week-Old Pigs from an Australian Pig Herd Using Metagenomic Sequencing. Viruses, 13(8), 1608. https://doi.org/10.3390/v13081608