1. Introduction
Bacteriophages (phages) are viruses that replicate in bacterial hosts and have been gaining attention for their potential use in biocontrol applications [
1,
2]. Phages are estimated to infect every bacterial genus, outnumbering bacteria by approximately tenfold [
3]. Despite the overwhelming number of phages present in the biosphere, very few studies provide detailed descriptions of phage dynamics with their hosts.
The genus
Acinetobacter has approximately 100 identified and 37 sequenced phages [
4]. The majority of these phages belong to the dsDNA
Caudovirales and infect the emerging pathogen
Acinetobacter baumannii. This species is at the top of the list provided by the World Health Organization and the Centers for Disease Control and Prevention describing pathogens for which new antimicrobials are most desperately needed [
5]. Thus, the majority of phage screens have been directed toward this pathogen, and the host range for these
Acinetobacter-infecting phages will be limited to
A. baumannii. Recently, we isolated the
Acinetobacter radioresistens strain LH6 from chicken feces on a free-range farm [
6]. We found that this multidrug resistant reservoir strain encodes genes associated with resistance to toxic metals and quaternary ammonium compounds [
7]. We also confirmed the presence of the infamous
bla oxacillinase gene, which has been directly linked to extreme drug resistance and is believed to originate from this species, although LH6 lacks the accessory insertional element necessary to induce expression of the gene [
7]. In addition, LH6 is capable of tolerating desiccation for significantly longer periods of time compared to
A. baumannii and
Escherichia coli [
8], with reports describing desiccation revival after >160 days and tolerance to radiation for this species [
9].
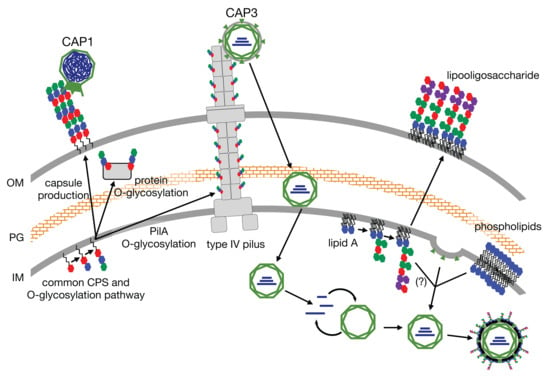
One contributing factor to the exceptional environmental stress tolerance of LH6, and
Acinetobacter strains more generally, are their diverse glycan structures [
10,
11]. Acinetobacters possess pathways for the biosynthesis of capsular polysaccharides (CPS), trehalose [
12] lipooligosaccharides (LOS), poly-N-acetylglucosamine (PNAG), and O-linked glycoproteins (modified with O-glycans) [
10], all of which can play significant roles in
Acinetobacter persistence and virulence [
10]. In
A. baumannii, CPS and O-glycans use a common pathway to assemble oligosaccharides onto the lipid carrier undecaprenylphosphate (UndP) in the inner leaflet of the inner membrane before being flipped into the periplasmic space and transferred to proteins (including pilin) or polymerized into CPS and exported to the outer membrane [
13,
14]. Similarly, the LOS is produced via the Raetz pathway, wherein Kdo
2-lipid A is built in the inner leaflet of the inner membrane and the oligosaccharide core is assembled sequentially onto the first Kdo sugar moiety, yielding LOS. The LOS is then flipped into the periplasmic space and transferred to the outer membrane via the Lpt pathway [
15], readily recognizable from the genomic sequence of LH6 [
6]. Another sugar structure that is commonly produced by acinetobacters is the PNAG polymer. PNAG is formed by polymerizing N-acetylglucosamine and releasing the polysaccharide into the extracellular matrix during biofilm assembly and to aid in adherence to surfaces [
16]. Together, all of these sugar structures contribute to the tenacity and pathogenesis of
Acinetobacter species.Along with the isolation of LH6, we conducted phage screens using fecal samples collected from poultry species housed on the same farm. We isolated several phages classifying into three different families, including an induced lysogenic syphovirus and two lytic podoviruses, all infecting strain LH6. Amongst these phages, we also isolated the first reported group of segmented dsRNA phages in a species outside of the genus
Pseudomonas. Before this report, there were seven sequenced pseudomonad dsRNA phages in the International Committee on Taxonomy of Viruses (ICTV) database constituting the family
Cystoviridae [
17]. Cystoviruses are viruses with a life cycle unique to phages, which has been studied extensively in the type phage Phi6 [
18]. In general, they are composed of a tripartite segmented dsRNA genome within two capsids enveloped in the inner membrane of the host in which they last replicated. After infecting through the pilus or rough lipopolysaccharide (LPS, or what we refer to as LOS) of the host, the dsRNA segments are polymerized while shielded by the incomplete capsid as polycistronic mRNA, limiting exposure of dsRNA, which is foreign to host nucleases and could be destroyed [
18]. After viral replication, the RNA segments are packed into assembled capsids where the genome is replicated by integral RNA polymerases. The phage capsid is then enveloped in the host inner membrane before degrading the peptidoglycan, resulting in lysis of the outer membrane and exiting out of the host cell (
Figure 1) [
17].
In this study, we sequenced all five of the isolated cystoviruses and selected one, CAP3, for further characterization. We were particularly interested in whether the host-derived envelope of CAP3 captured any glycan intermediates known to be actively assembled at the inner membrane into the diverse array of glycan structures that are subsequently exported in the host. To date, no host-associated structural glycoconjugates have been described for these or other small bacteriophages. Knowing that the membranes of Acinetobacter are rich in lipid-linked glycans we hypothesized that the phage would coat itself in host membranes containing these glycoconjugates. We also mutated select LH6 glycan biosynthesis gene homologs to assess their impact on phage infection and potential envelope modification. Together, these results describe three diverse and one novel Acinetobacter-infecting phage genera with several unexpected features requiring further study.
2. Materials and Methods
2.1. Bacterial Growth Conditions
A. radioresistens strain LH6 was grown aerobically using Luria–Bertani (LB) medium (Becton, Dickinson and Company, Franklin Lakes, NJ, USA) at 30 °C and under agitation at 200 rpm for liquid culture. In phage propagation conditions, brain heart infusion (BHI) medium (Hardy Diagnostics, Santa Maria, CA, USA) was used to increase yields.
2.2. Lytic Bacteriophage Propagation and Isolation
Bacteriophage isolation was performed as described [
19]. Propagating strain LH6 was grown overnight at 30 °C with shaking at 200 rpm, the culture was adjusted to OD
600 = 0.3, and infected with bacteriophages at a multiplicity of infection (MOI) of 0.001. The infected culture was incubated at 30 °C with shaking at 200 rpm overnight. Afterward, the culture was centrifuged at 4255×
g for 15 min, the resulting supernatant was filtered through a 0.22 μm filter and the phage-containing filtrate was collected.
2.3. Lysogenic SLAP1 Bacteriophage Induction and Isolation
For SLAP1 prophage induction, propagating strain LH6 was grown in BHI broth overnight at 30 °C, shaking at 200 rpm. The culture was adjusted to OD
600 of 0.05 and was grown overnight in the presence of 1 µg/mL mitomycin C (Gold-Biotechnology, St. Louis, MO, USA). After centrifugation at 7020×
g for 10 min, the phage-containing supernatant was filtered (0.2 µm), and the phage-containing filtrate was collected. The filtered supernatant was then ultra-centrifuged at 141,000×
g for 1.5 h at 4 °C. The resulting phage pellet was resuspended in SM buffer (100 mM NaCl, 8 mM MgSO
4•7H
2O, 50 mM Tris-HCl pH 7.5, 0.002% (
w/
v) gelatin). For high-resolution transmission electron microscopy (TEM) imaging, the concentrated prophages were treated with chloroform. Briefly, 0.1 volume of chloroform was added to the phage solution, vortexed occasionally and incubated for 10 min at RT. The suspension was centrifuged at 4000×
g for 5 min, the top layer containing the phages was transferred to a new tube and imaged as described below. For the clearance assay, this was done similarly to the spot assay described previously [
7]. Briefly, the host strain LH6 was grown overnight and adjusted to OD
600 = 0.3. Then, 500 µL bacteria, 5 mL of molten BHI 0.6% agar, and in the case of SLAP1, 1 µg/mL mitomycin C, were mixed and poured onto a BHI agar plate. After the agar solidified, 10 µL of the phage suspension was spotted onto the medium and the plate was incubated overnight.
2.4. Bacteriophage Transmission Electron Microscopy
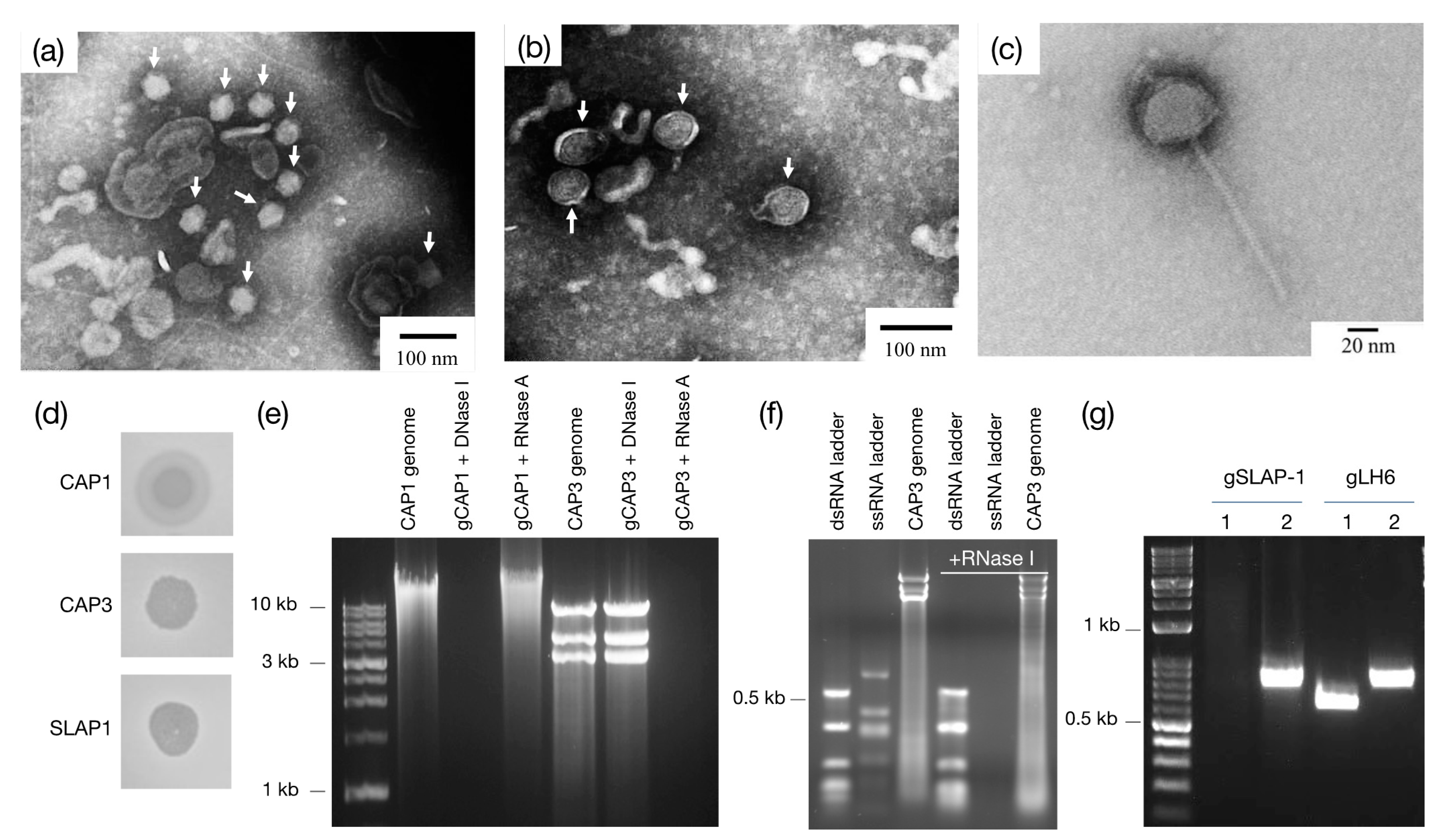
Following propagation, bacteriophages CAP1 and CAP3 were incubated for 5 min at room temperature with host strain LH6 (OD600 = 0.5) at MOI of ~100. Similarly, 50 µL of concentrated SLAP1 was incubated for 2 min with 250 µL LH6 cells at OD600 = 0.5. Then 5% paraformaldehyde (Electron Microscopy Sciences, Hatfield, PA, USA) was added to fix the samples and halt the infection. The samples were spotted onto parafilm and formvar-coated copper grids (Electron Microscopy Sciences) were placed on top of each drop and left to incubate for 1 h at room temperature, followed by three 3 min washes in each 1x PBS and ddH2O. The grids were negatively stained with 0.5% phosphotungstic acid (Electron Microscopy Sciences) for 10–30 s, wicked onto a Kimwipe and imaged with the JEOL JEM1011 TEM (JEOL Inc., Akishima, Tokyo, Japan).
2.5. Genome Characterization
Phage genomes were isolated by the phenol/chloroform method described [
20]. To determine the nucleic acid composition, 1 μg of each genome was incubated with DNase I or RNase A (ThermoFisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions, separated on a 1% agarose gel and imaged. To determine ds or ssRNA in CAP3, RNase I
f (New England BioLabs, Ipswich, MA, USA) was added per manufacturer’s instructions to the ssRNA ladder (New England BioLabs), dsRNA ladder (New England BioLabs) and CAP3 genomic RNA. The reaction was stopped with 0.1% sodium dodecyl sulfate (final volume) and separated on a 2% agarose gel and imaged.
2.6. CAP3–CAP7 Whole Genome Sequencing
Phage genomes were provided to GENEWIZ for cDNA library construction and Illumina sequencing using 150 bp reads with >100x coverage. Illumina reads were quality filtered (≥Q30) using Geneious Prime (v2020.1), and then high-quality filtered reads were mapped to the A. radioresistens LH6 genome to eliminate any reads from rRNA loci or other LH6 gene expression. Next, unmapped reads were assembled into contigs using Newbler assembler (v2.6), and the resulting contigs were cleaned and the finalized chromosome assembly for CAP3–7 was done using Geneious Prime.
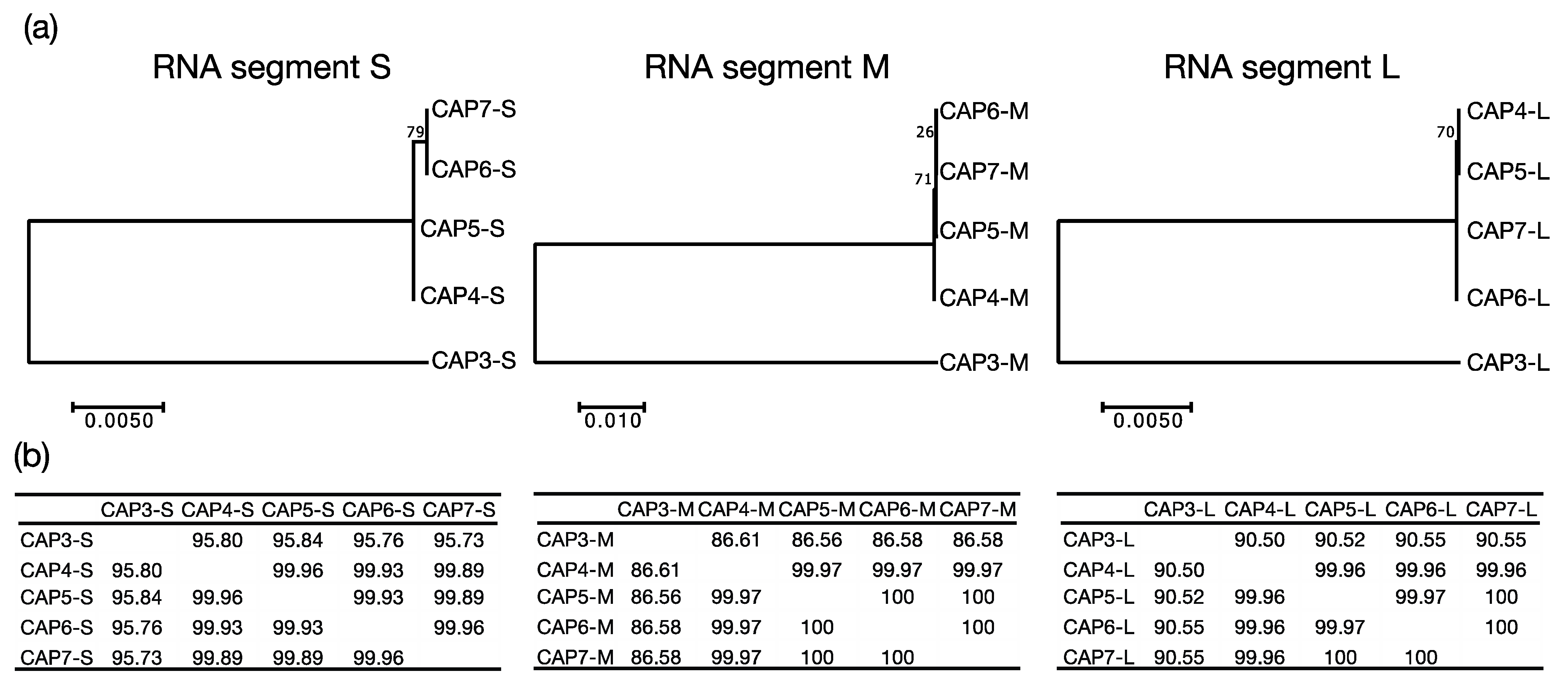
The evolutionary history was inferred by using the Maximum Likelihood method based on the Kimura 2-parameter model [
21]. The trees with the highest log likelihood (segment 1: −10401.29, segment 2: −4365.84, segment 3: −6612.83) are shown. The percentage of trees in which the associated taxa clustered together are shown next to the branches. Initial tree(s) for the heuristic search were obtained automatically by applying Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Maximum Composite Likelihood (MCL) approach, and then selecting the topology with superior log likelihood value. The trees are drawn to scale, with branch lengths measured in the number of substitutions per site. The analysis involved 5 nucleotide sequences. There were a total of 6614 positions (base pairs) for segment 1, 2738 positions for segment 2, and 3666 positions for segment 3 in the final dataset. Evolutionary analyses were conducted in MEGA7 [
22].
2.7. Identification of Induced Prophage SLAP1
For the identification of the induced prophage SLAP1, primers were designed that target the capsid genes of prophage 1 and prophage 2 [
6]. Prophage 1 was detected with the primers pP1capsid-F and pP1capsid-R, prophage 2 was detected with the primers pP2capsid-F and pP2capsid-R (primers listed in
Table S1). The PCR was performed with OneTaq polymerase (New England BioLabs) following the manufacturer’s protocol with the annealing step at 52 °C for 1 min and the elongation at 68 °C for 45 s. As template, 130 ng SLAP1 and LH6 genome were used, isolated via phenol/chloroform extraction and E.Z.N.A Bacterial DNA kit (Omega Bio-tek, Inc., Norcross, GA, USA), respectively. The PCR fragments were cleaned up via the DNA Clean & Concentrator-25 Kit (Zymo Research Inc., Irvine, CA, USA) and run on a 1% agarose gel for visualization.
2.8. Mutagenesis of LH6
The ∆
pglC (DOM24_00295) knockout construct was made in pGEM-t-easy (Promega, Madison, WI, USA), with ~1000 bp homology upstream and downstream of
pglC flanking either side of the kanamycin cassette from the plasmid pBAV1K-T5-
gfp [
23]. The primers (
Table S1) used to generate the flanking regions were: ApaI-KO
pglC upstream-F, SphI-KO
pglC upstream-R, SpeI-RBS-KO
pglC downstream-F, and PstI-KO
pglC downstream-R. The ∆
pilA (DOM24_13175) knockout construct was made in the same plasmid vector as for
pglC. The ~900 bp upstream
pilA mutant construct was fused with the kanamycin cassette promoter to ensure the function of the upstream gene, the primers used for this fragment were: ApaI-KO
pilA upstream-F, NcoI-KO
pilA upstream-R, KO
pilA upstream with kan pro-F, and KO
pilA upstream with kan pro-R. The primers for the ~1000 bp homology downstream were: SalI-KO
pilA downstream-F, and SacI-KO
pilA downstream-R. The ∆
lpsC (DOM24_02210) and
clsB (DOM24_06510) knockout constructs were made in the same plasmid vector. In the
lpsC knockout construct, the ~1000 bp homology upstream and downstream were made by primers: ApaI-KO
lpsC upstream-F, SphI-KO
lpsC upstream-R, SpeI-RBS-KO
lpsC downstream-F, and SalI-KO
lpsC downstream-R. In the ∆
clsB knockout construct, the ~1000 bp homology upstream and downstream were made by primers: SphI-KO
clsB upstream-F, NcoI-KO
clsB upstream-R, SpeI-RBS-KO
clsB downstream-F, and SalI-KO
clsB downstream-R. After construction of the knockout constructs, LH6 cells were mutated with an adaptation of the previously described recombineering method in
A. baumannii [
24]. Briefly, the plasmid pAT4 was inserted into LH6 and induced for four hours with 2 mM IPTG to express RecAB. The cells were then washed 3x with 10% glycerol and electroporated with a BioRad GenePulser on the Ec2 setting. The electroporated cells were then grown in 4 mL of liquid culture with 2 mM IPTG for 2 h before collecting the cells and plating on LB+ kanamycin (50 µg/mL) to select for mutants. The isolated mutants were then confirmed by PCR (
Figure S1) and Sanger sequencing.
2.9. Purification of CAP Phages
Phages were propagated as described above on LH6 WT, ∆pglC, ∆lpsC and ∆clsB and the phage filtrate was collected. The phage was then purified with poly(ethylene glycol) 8000 (PEG 8000) precipitation and cesium chloride (CsCl) step-gradient protocols, adapted for use with these phages. The phage filtrate was incubated with 25 U/mL Turbonuclease (Sigma Aldrich, St. Louis, MO, USA) at room temperature for 30 min. Sodium chloride was added to a final concentration of 1 M, and the phage suspension was cooled in ice water for 1 h. Then, PEG 8000 was added to a final concentration of 10% and was dissolved with slow stirring. The phage suspension was transferred to centrifuge bottles and incubated at 4 °C overnight to allow phages to precipitate. After precipitation, phages were pelleted by centrifugation at 11,300× g for 25 min. The supernatant was removed and 5 mL sterile SM buffer (without gelatin) was added to saturate the pellets for 1 h at room temperature. The pellets were then resuspended and transferred to a sterile 15 mL tube for storage before purification in the CsCl step-gradient.
The CsCl step-gradient was prepared by making CsCl solutions in sterile SM (without gelatin) at the following concentrations: 1.3 p = 0.404 g/mL; 1.5 p = 0.675 g/mL; 1.7 p = 0.943 g/mL. The layers were created in a Beckman SW-28 ultracentifuge polycarbonate insert tube by adding the least dense layer first and injecting denser layers on the bottom with a long needle attached to a syringe. The PEG-purified phages were added on top of the CsCl gradient and ultracentrifuged for 2.5 h at 25,000 rpm (~83,000× g) at 4 °C. After the phages were separated from impurities (appearing in the gradient as a blue-white band), the sidewall of the tube was punctured by an 18-gauge needle and the band was extracted. The phage solutions were then dialyzed using 3500 MWCO dialysis tubing in SM (without gelatin) overnight at 4 °C. The dialyzed phages were transferred to fresh SM for two additional hours before being removed from the dialysis tubing and filtered (0.2 µm), and were then stored at 4 °C.
2.10. SDS-PAGE Analysis of Phage Glycans
Samples were standardized by protein content using a Pierce™ BCA kit (ThermoFisher) for CsCl-purified phages and by OD600 for bacterial cells, mixed with 5× SDS-loading dye (0.25% (w/v) bromophenol blue, 0.5 M DTT, 50% glycerol, 10% (w/v) SDS, 0.25 M Tris-HCl pH 6.8, 5% 2-mercaptoethanol) to a final concentration of 1×, heated at 95 °C for 15 min, and then loaded onto the 12.5% or 15% SDS-PAGE gels. Where indicated, proteinase K was added to a final concentration of 1 mg/mL and incubated with the samples for 18 h at 37 °C, followed by heating at 95 °C for 15 min and cooling to room temperature before loading onto the gels. Where indicated, glacial acetic acid was added to a final concentration of 1% and heated to 95 °C for 30 min. Proteins were separated by first running at 100 V for 10 min and then 170 V for 80 min. Samples were stained with the Pro-Q™ Emerald 300 (ThermoFisher) glycan staining kit according to manufacturer’s instructions. The same samples were also prepared without proteinase K treatment, separated on SDS-PAGE gels and stained with Coomassie stain.
2.11. Glycosyl and Fatty Acid Composition Analysis of CAP3 and LH6 by Gas Chromatography-Mass Spectrometry (GC-MS)
CsCl-purified CAP3 and LH6 WT cells were prepared for glycosyl and fatty acid compositional analysis by first proteinase K treating (0.5 mg/mL) for 24 h at 37 °C, followed by 1 h at 60 °C. The proteinase K treated lysates were then dialyzed with a 1000 MWCO dialysis tubing into mqH2O before being lyophilized. Ten milligrams of cell or phage material was then dissolved into mqH2O and an equal volume of chloroform was added. The solution was vortexed for 30 s and phases were separated by centrifugation for 5 min at 4000× g. The aqueous phase was collected and an equal volume of fresh water was added; the process was repeated until three aqueous phases were collected. Then, the organic phase was collected in a similar manner for a total of three fractions. The interphase was collected last by dissolving in a small volume of water and chloroform. The water, chloroform and interphase samples were evaporated to dryness and the compositional analysis was performed by preparing trimethylsilyl (TMS) methyl glycoside derivatives after 18 h of methanolysis with 1 M HCl-MeOH at 80 °C. In addition to glycosyl composition, the methanolysis also allowed the conversion of the fatty acids to methyl esters. To detect amino sugars, the hydrolyzed samples were re-N-acetylated (methanol: pyridine: acetic anhydrate; 2:1:1 vol. at 100 °C; 1 h) and all methyl glycosides and methyl esters were finally converted into TMS-methyl glycosides and hydroxyl-FAMES (Fatty Acid Methyl Esters) into TMS-FAMES by using Tri-Sil HTP (Thermo) reagent for 30 min at 80 °C. The TMS derivatives were analyzed by GC-MS on a Hewlett-Packard HP5890 gas chromatograph equipped with a mass selective detector 5970 MSD using EC-1 fused silica capillary column (30m × 0.25 mm I.D.), and temperature program at 80 °C for 2 min, then ramped to 160 °C at 20 °C/min with a 2 min hold, and to 200 °C at 2 °C/min followed by an increase to 300 °C at 10 °C/min with a 20 min hold. The fatty acid identity was assigned based on the unique electron ionization (EI)-MS fragmentation patterns of the FAME of straight-chain (saturated and unsaturated) fatty acids and the EI-MS fragments of the FAME-TMS of hydroxylated fatty acids.
2.12. DOC-PAGE Analysis of Bacterial and Phage Extracts
The CAP3 and LH6 aqueous, interphase, and organic extract phases were resolved by PAGE by using 18% acrylamide and deoxycholic acid (DOC) detergent [
25]. The gels were stained with silver using the Bio-Rad Silver Staining Kit (Bio-Rad, Hercules, CA, USA). In addition, the gels were stained with Alcian blue dye [
26], followed by the same steps as with standard silver staining.
4. Discussion
Bacteriophages are predicted to infect every bacterial genus, but scientists have only examined the tip of the iceberg when exploring the diversity of phages that are all around us. In this work, we describe the isolation and characterization of eight phages infecting the multidrug resistant
A. radioresistens strain LH6. CAP1 and CAP2 were isolated from chicken feces, while CAP3 was from duck feces, and CAP4–7 were isolated from turkey feces. Finding phages infecting LH6 that were isolated from the same chicken feces, is not surprising, however, the isolation of CAP3–7 from different birds was unexpected. MEGA7 phylogenetic analysis grouped CAP3 apart from CAP4–CAP7 (
Figure 3), which is suggestive of a persistence in the duck-husbandry environment, separate from the turkeys that CAP4–CAP7 were derived from. The extremely high sequence similarity of the CAP4–CAP7 phages suggests that they are likely the same phage, or variants of the same phage. The background rate of mutation and genetic drift for these phages is unknown, but this information could yield insight into how recently the phages shared an ancestor. Our results suggest that either a compatible strain of
A. radioresistens was present in those birds, which the CAP phages could infect and replicate in, or these phages have a high environmental persistence in a poultry-husbandry setting.
The different plaque morphologies of the isolated phages are also of interest. CAP1 and CAP2 both have a “halo” phenotype surrounding the observed plaques (
Figure 2d). This can be explained by diffusion of a phage-associated glycolytic enzyme, and is consistent with other bacteriophages [
28], including
Salmonella-infecting phage P22 which enzymatically degrades the
Salmonella O-antigen polysaccharide [
29]. CAP3–7, however, have a different plaque morphology which represents the more classic “pinprick” type plaque with a defined zone of clearing. SLAP1 is different from either of the other plaque morphologies. The zone of clearing produced by spotting the phage onto LH6 is semi-transparent, probably due to the lysogenic nature of the phage which allows for the growth of LH6 even in the presence of high phage concentrations. Interestingly, the phage can only be visualized by plaquing assays in the presence of mitomycin C, without which, no zone of clearing is formed, even at high phage densities (results not shown).
After genome and TEM characterization of the CAP1–7 and SLAP1 phages, we propose that CAP1 and CAP2 belong to the
Podoviridae. The DNA content, icosahedral heads and short tails are consistent with other members of this phage family [
30]. Second, we propose that SLAP1 belongs to the
Siphoviridae due to the icosahedral head and long flexible tail, which is the hallmark of this phage family [
31]. Finally, we propose that CAP3–CAP7 belong to a new phage genus within the family
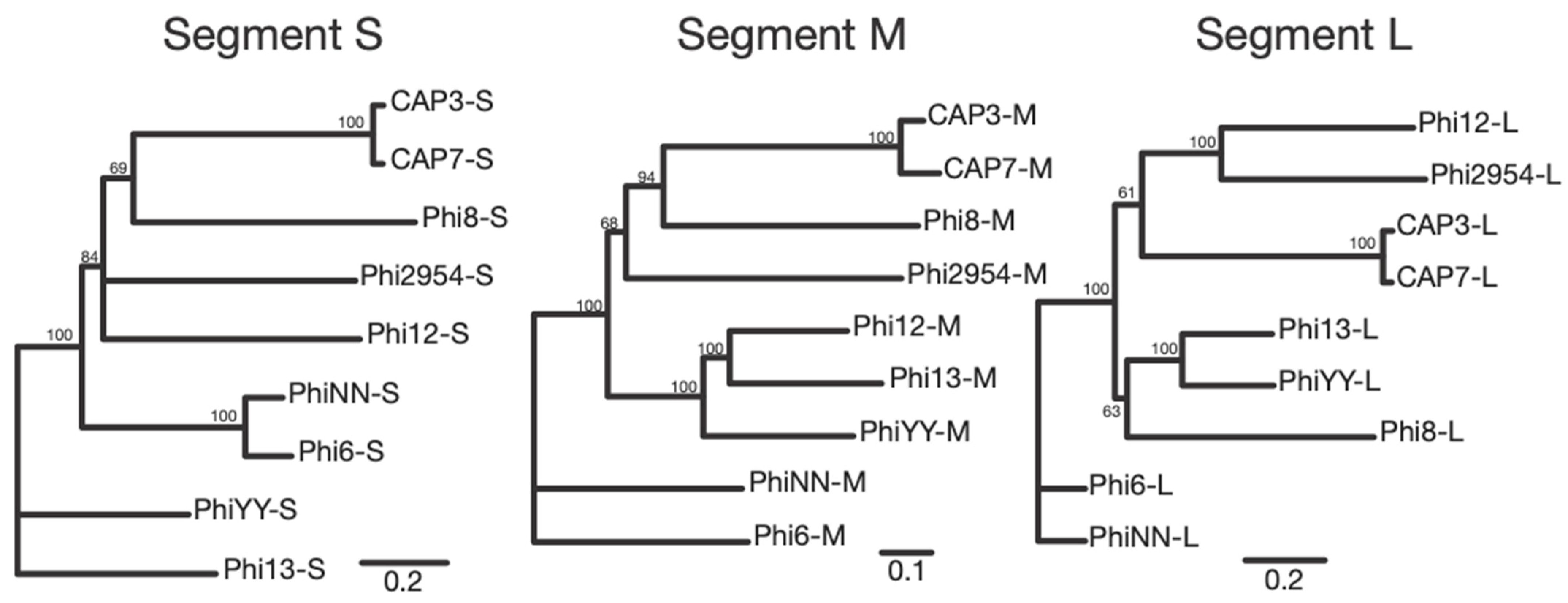
Cystoviridae. The enveloped capsid, along with the segmented dsRNA genome are indicative of this family, along with the sequencing identity, which shares synteny and some homology with other cystoviruses infecting members of the
Pseudomonas genus (
Figure 6 and
Figures S2–S4) [
32,
33]. Interestingly, these are the first cystovirus phages that infect any species besides the seven characterized phages that infect
P. syringae and
P. aeruginosa [
17]. Whole genome sequencing and subsequent analysis showed that some of the CAP genes could be assigned putative functions based on synteny to the other cystoviruses (
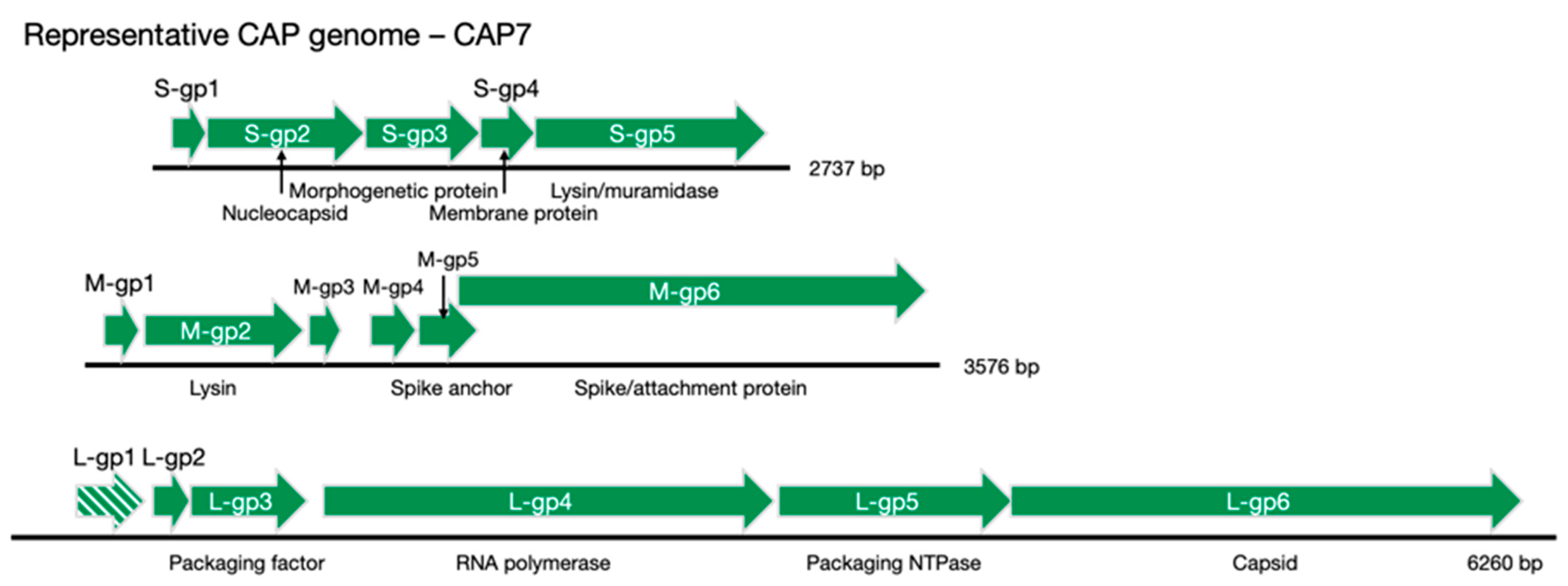
Table 1,
Figure 3). The lack of distinct synteny shows that these viruses mutate at a high rate and can undergo genomic rearrangement, while maintaining a similar genome size and life cycle. Better understanding of these processes could give insight into the evolutionary tendencies of RNA viruses as whole.
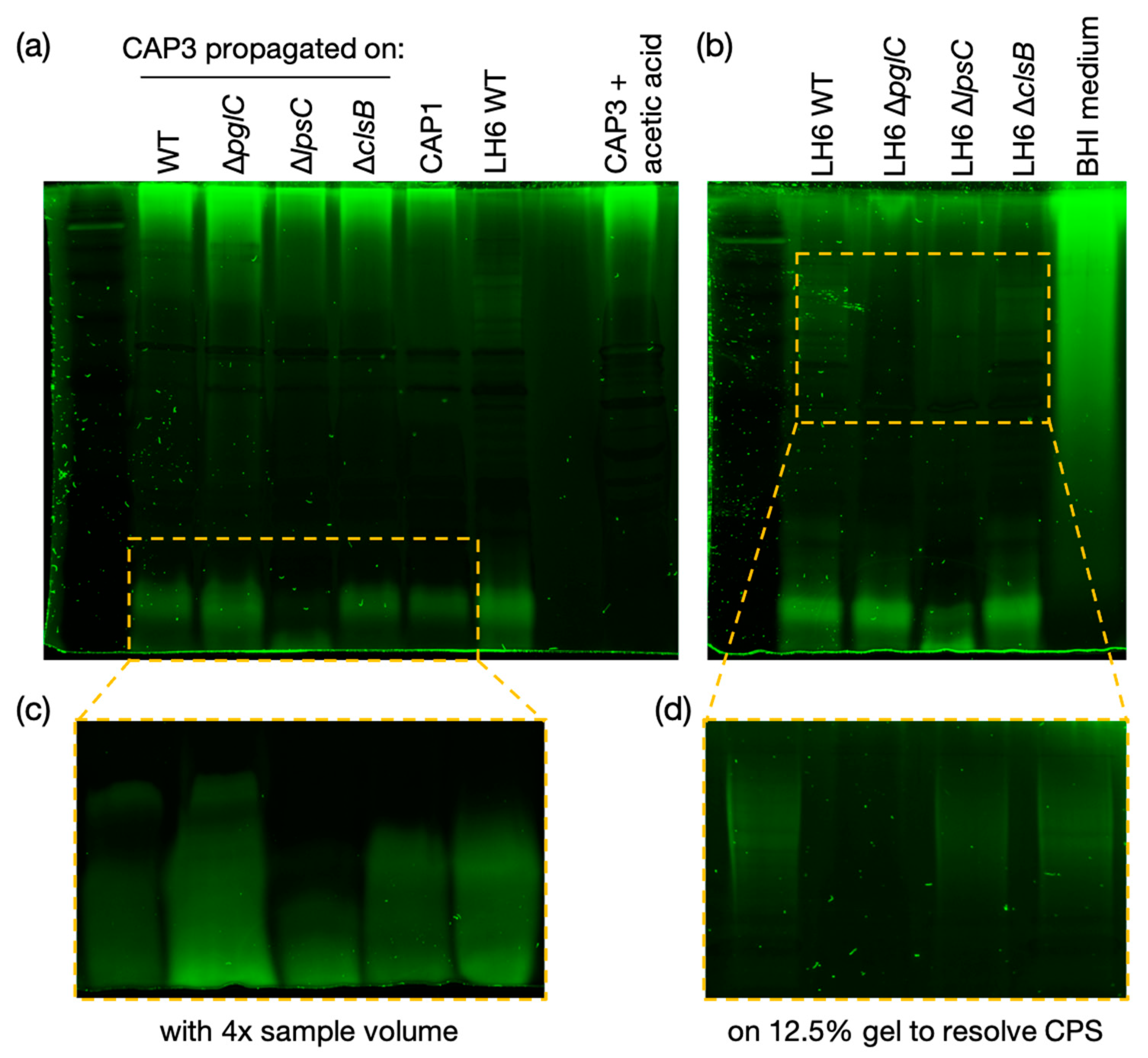
After identifying CAP3–7 as cystoviruses, we were interested in investigating whether or not their envelopes, derived from the host’s inner membrane [
32], contained any glycan intermediates known to be synthesized at this location. To test this, we performed several glycan stains on CAP3 lysates and observed two areas of SDS-PAGE staining corresponding to a high MW glycan and a low MW glycan. We determined that the high MW stained material was a contaminant from the growth medium while the lower MW structure is a lipid-linked glycan, presumably an oligosaccharide attached to lipid A (i.e., LOS) in
A. radioresistens. Using mutants disrupting CPS/O-glycan, LOS core and cardiolipin (or trehalose phospholipid [
34]) biosynthesis, we were able to determine that the low MW glycan is not a precursor to CPS, and was similarly unaffected in the ∆
clsB mutant strain and derived-phage samples. The glycoconjugate was, however, truncated after the LH6 mutation of
lpsC and subsequent phage propagation on that strain. LpsC is homologous to conserved LOS core β-glucosyltransferases found in most Gram-negative bacteria [
35]. Inactivation of the enzyme leads to LOS truncation, consistent with the faster LOS migration observed by SDS-PAGE for both LH6 ∆
lpsC and CAP3 propagated on this mutant. Similarly, mass-spectrometry analyses of both LH6 and purified CAP3 showed signals corresponding to β-hydroxy fatty acids unique to bacterial lipid A (
Figure 8). However, it remains to be determined whether cystoviruses have glycoconjugates (i.e., host lipooligosaccharides) embedded in their membranes or whether our observations are an artifact of phage propagation on the host.
It is difficult to imagine a scenario where host glycan intermediates would not be present during phage induced membrane-budding, but cystovirus recruitment of host membranes appears to exclude host proteins [
36], so it is possible that only phospholipids are selectively recruited from the host. Laurinavičius et al. demonstrated that the Phi6 phage had a similar phospholipid content as its host’s inner membrane, and our GC-MS profiles also detect similar fatty acid compositions (16:0 and 18:1) [
37]. However, the researchers used organic extraction to intentionally isolate phospholipids while we observed that most CAP3 membrane components partitioned into the aqueous phase. Similar studies need to be done to determine the full composition of the LH6 inner membrane versus the LH6 host-derived membrane. In the Laurinavičius et al. study, the predominant lipids were phosphatidylethanolamine and phosphatidylglycerol, along with low levels of cardiolipin [
37]. Recent studies in
Salmonella have identified a new class of glycolipids, 6-phosphatidyltrehalose and 6,6-diphosphatidyltrehalose, that require the cardiolipin synthase (ClsB) for synthesis [
34]. Since we recently demonstrated that
A. radioresistens LH6 is capable of synthesizing very high levels of trehalose [
37], we mutated its
clsB homolog to see if any changes were observed in the CAP3 glycan staining profile (particularly since GC-MS analyses identified high levels of Glc in the CAP3 chromatogram,
Figure 8a), but again, a more thorough membrane compositional analysis of both LH6 and CAP3 needs to be done at a larger scale to confirm which glycoconjugate structures are actually present in the membrane; characterization of phage particles by immunogold labeling is currently underway to prove if any of these structures are phage-associated.
Additionally, we were interested in identifying the phage receptors necessary for host recognition. Based on the halo plaque morphology observed for CAP1, we predicted this phage would interact with a surface polysaccharide, and since Acinetobacters, including other strains of
A. radioresistens [
38], typically express rough LPS (i.e., LOS), we wanted to target the CPS. We identified several putative CPS transport and biosynthesis genes including
pglC, which is predicted to encode an enzyme catalyzing the addition of di-N-acetylbacillosamine (diNAcBac) or GlcNAc to the lipid carrier UndP based on gene homology and synteny with the related
A. baumannii [
14,
39,
40]. We presume that the O-linked glycosylation pathway is similarly impeded (
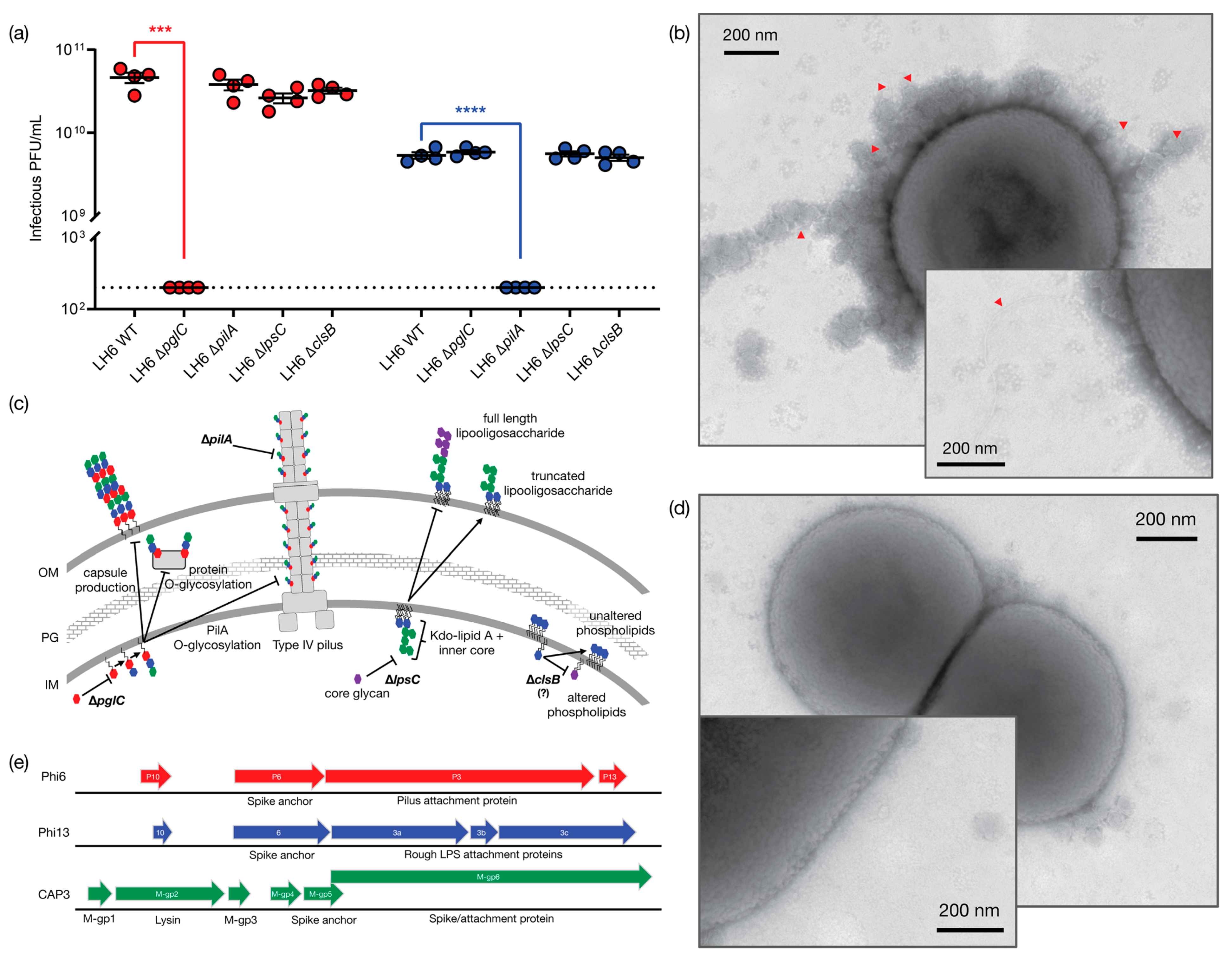
Figure 3b), but have not yet confirmed this experimentally. We then tested the ability of the representative phages CAP1 and CAP3 to plaque on the ∆
pglC mutant (
Figure 3a) and as predicted, CAP1 was unable to plaque on this mutant. We next sought to identify the host receptor for CAP3. The best-characterized member of the cystovirus family, Phi6, binds to the pilus of
P. syringae [
41], while other cystovirus phages bind to LPS. The high sequence synteny of the CAP3 M segment to the pilin-binding phage Phi6, is suggestive that the M-gp6 gene encodes a protein that will form a trimeric spike protein mediating host attachment. Furthermore, in other cystoviruses that do not bind pilin, but instead target LPS, the spike homolog is split into three ORFs that form a heterotrimer to bind
Pseudomonas LPS (
Figure 3c,
Figure S3). We thus hypothesized that CAP3 would require PilA and this was supported by our experiments demonstrating that CAP3 did not plaque on the LH6 ∆
pilA mutant, indicating that CAP3 requires the structural component of the LH6 pilus. Interestingly, it was recently shown that when the
P. syringae pilA gene was expressed in a
P. aeruginosa ∆
pilA mutant, both the
P. syringae phage Phi6 and the
P. aeruginosa phage PO4 were capable of infecting
P. aeruginosa [
42], suggesting that cystoviruses may be engineered to acquire a broader host range that extends beyond the strain and species level. These experiments represent the first step in understanding and potentially exploiting these
Acinetobacter viruses to recognize and infect other notorious
Acinetobacter species, which result in some of the most deadly and difficult to treat bacterial infections in the world.


 ,
, 









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}