
Influenza Antigens NP and M2 Confer Cross Protection to BALB/c Mice against Lethal Challenge with H1N1, Pandemic H1N1 or H5N1 Influenza A Viruses


Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design
2.2. Animals
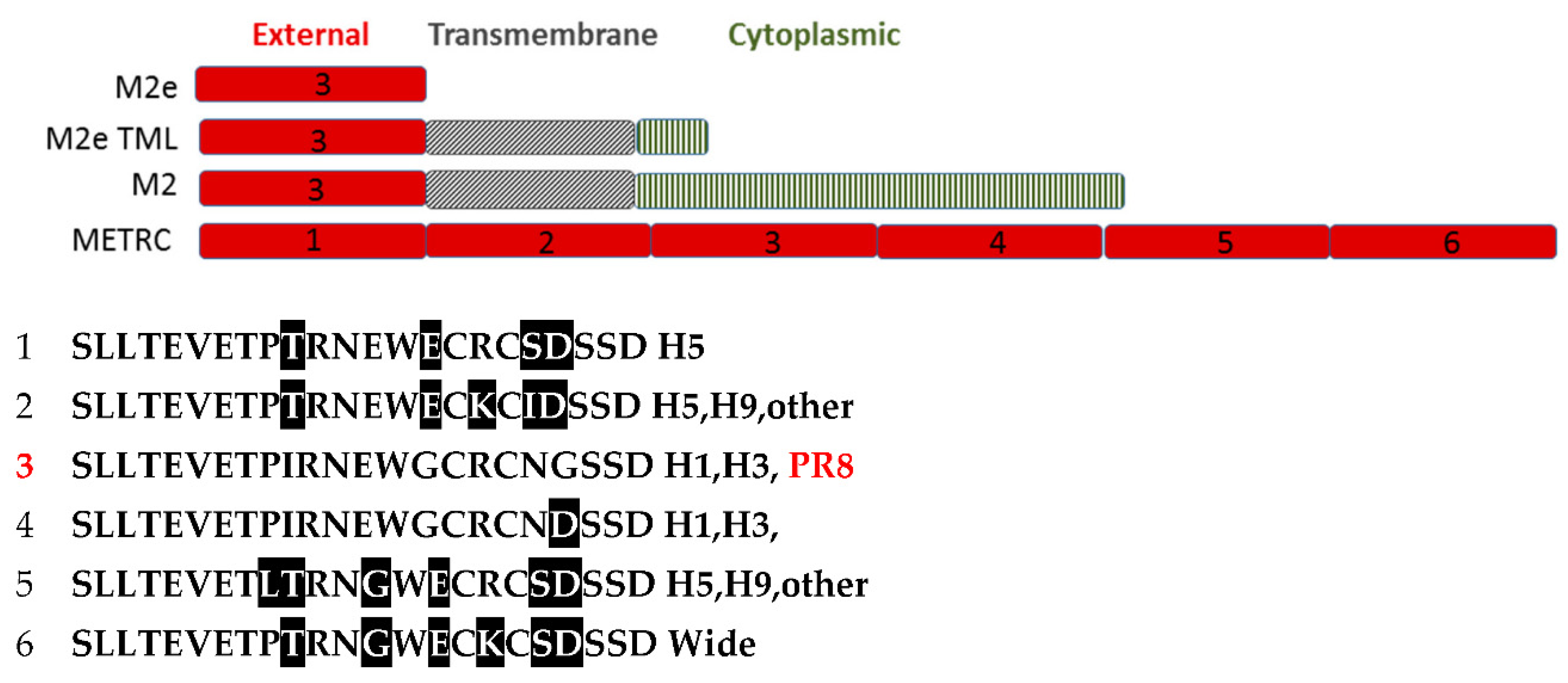
2.3. Vaccine Preparations
2.4. Challenge
2.5. Recombinant Proteins for ELISA
2.6. Hemagglutination Inhibition (HAI) Assay
2.7. Tissue Culture Infective Dose 50% (TCID50) and Viral Burden
2.8. Enzyme-Linked Immunosorbent Assays (ELISA)
2.9. Enzyme-Linked Immunospot (ELISpot) Assay
2.10. Ethical Approval
3. Results
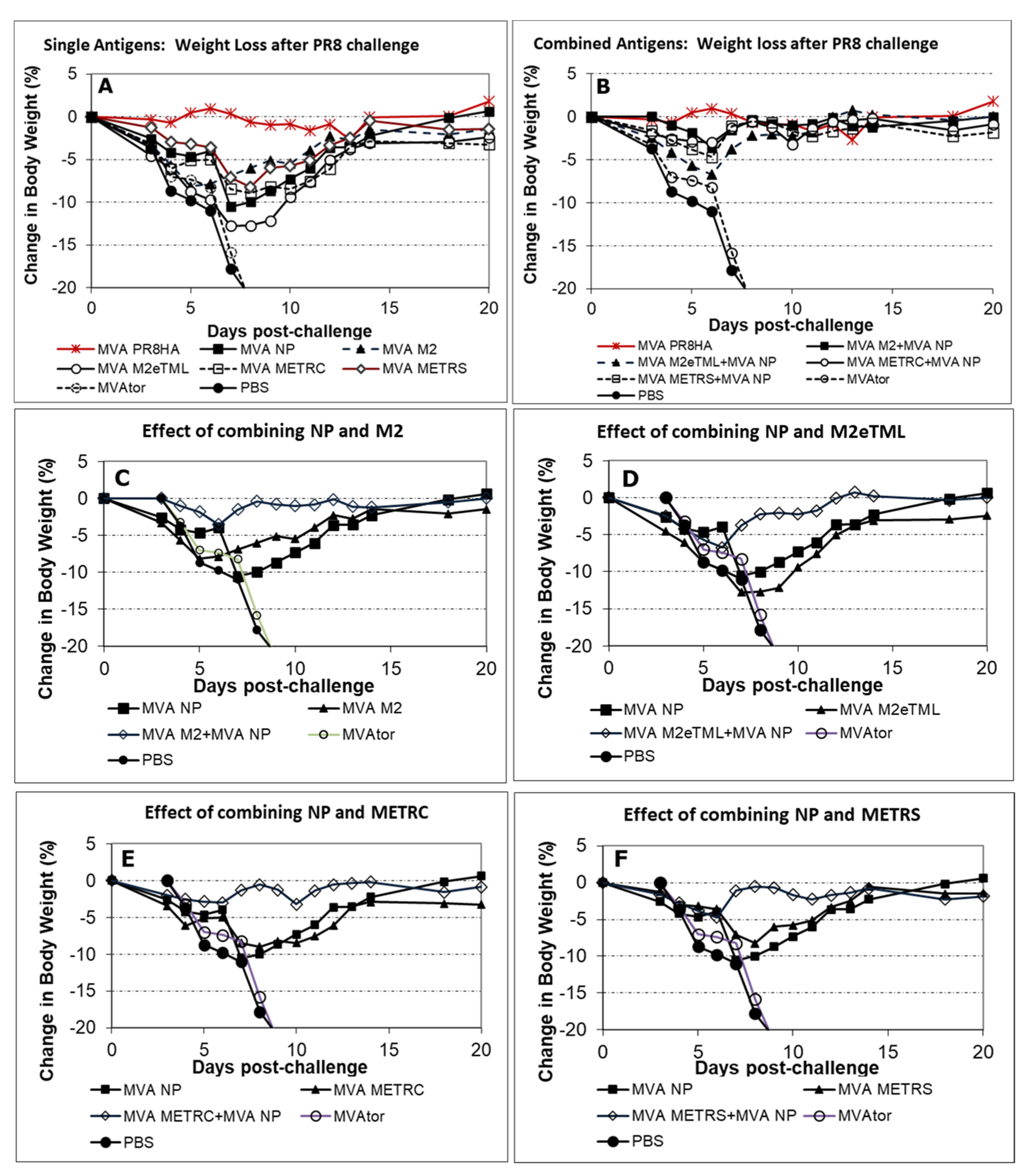
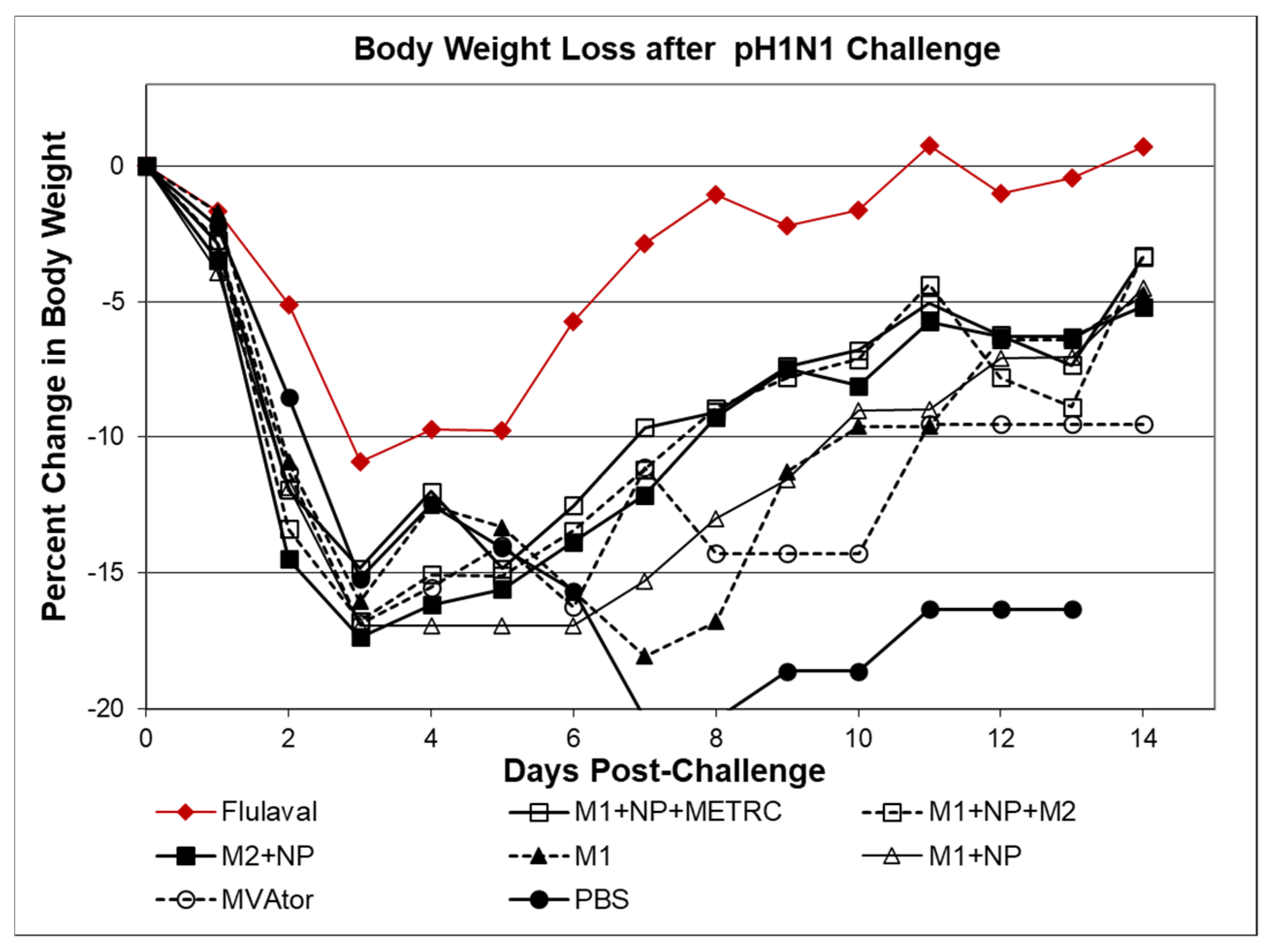
3.1. Survival and Body Weights Following INFV A Challenge
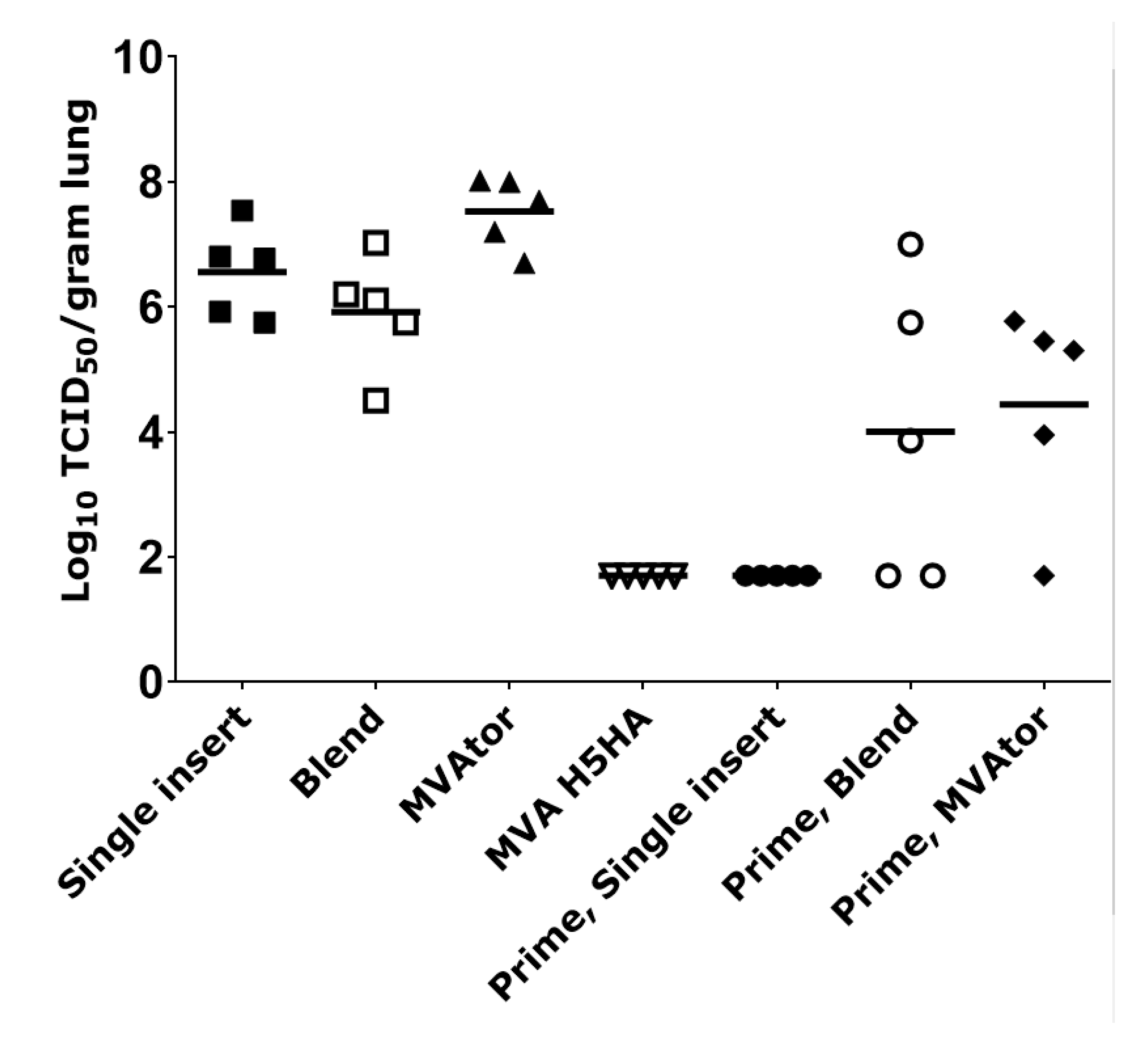
3.2. Viral Burden in Tissues
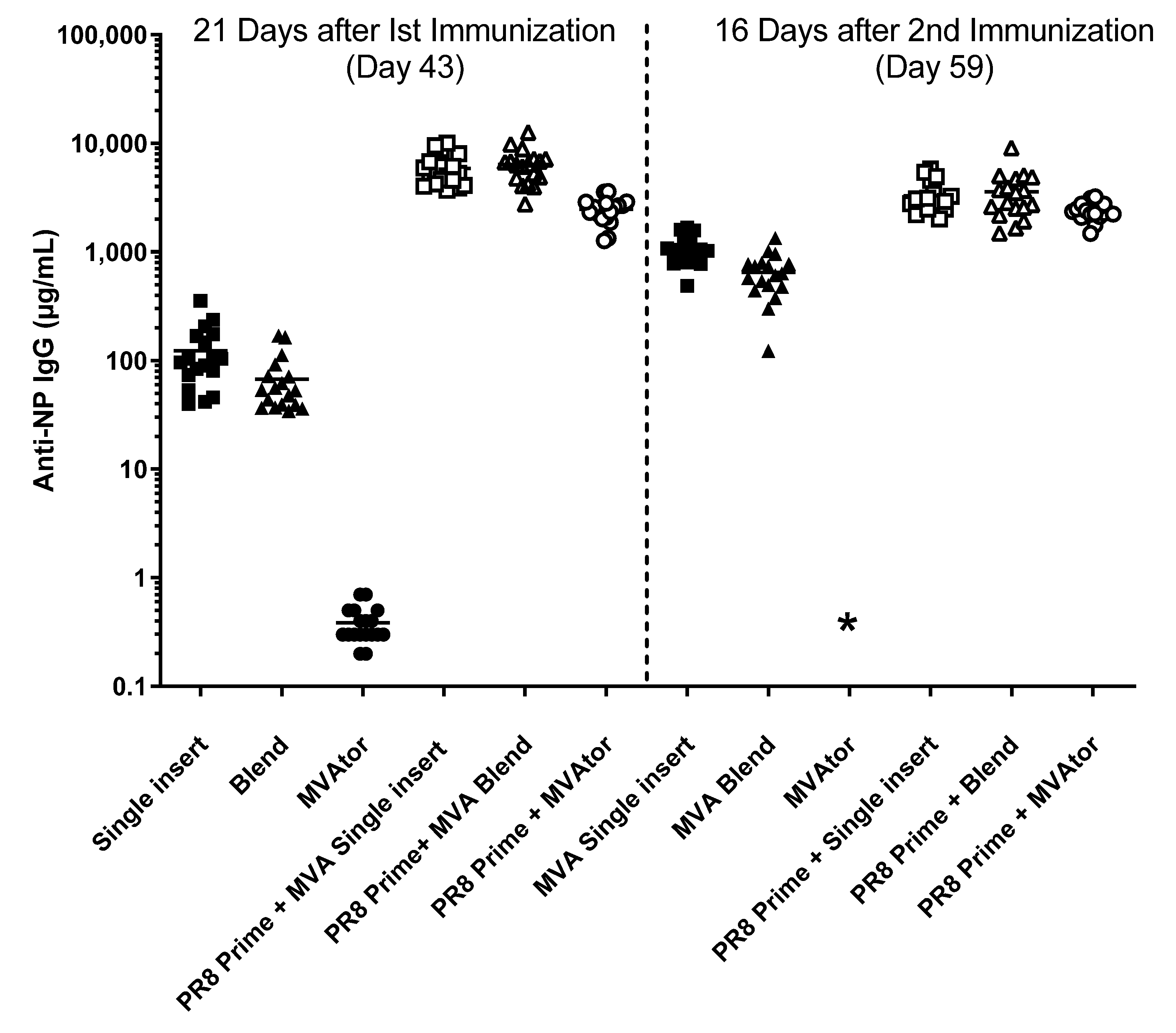
3.3. Antibody Responses to HA, NP, M2, M2e and M1 Antigens
3.4. MHC Class I and Class II-Restricted T Cell Responses
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Drake, J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef] [Green Version]
- Osterholm, M.T.; Kelley, N.S.; Sommer, A.; Belongia, E.A. Efficacy and effectiveness of influenza vaccines: A systematic review and meta-analysis. Lancet Infect. Dis. 2012, 12, 36–44. [Google Scholar] [CrossRef]
- Xie, H.; Li, X.; Gao, J.; Lin, Z.; Jing, X.; Plant, E.; Zoueva, O.; Eichelberger, M.C.; Ye, Z. Revisiting the 1976 "swine flu" vaccine clinical trials: Cross-reactive hemagglutinin and neuraminidase antibodies and their role in protection against the 2009 H1N1 pandemic virus in mice. Clin. Infect. Dis. 2011, 53, 1179–1187. [Google Scholar] [CrossRef] [Green Version]
- Glinsky, G.V. Genomic analysis of pandemic (H1N1) 2009 reveals association of increasing disease severity with emergence of novel hemagglutinin mutations. Cell Cycle 2010, 9, 958–970. [Google Scholar] [CrossRef] [Green Version]
- Welman, M.; Arora, D.J. Genomic analysis of matrix gene and antigenic studies of its gene product (M1) of a swine influenza virus (H1N1) causing chronic respiratory disease in pigs. Virus Genes 2000, 21, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Bean, W.J.; Schell, M.; Katz, J.; Kawaoka, Y.; Naeve, C.; Gorman, O.; Webster, R.G. Evolution of the H3 influenza virus hemagglutinin from human and nonhuman hosts. J. Virol. 1992, 66, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- Shu, L.L.; Bean, W.J.; Webster, R.G. Analysis of the evolution and variation of the human influenza A virus nucleoprotein gene from 1933 to 1990. J. Virol. 1993, 67, 2723–2729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerhard, W.; Mozdzanowska, K.; Zharikova, D. Prospects for universal influenza virus vaccine. Emerg. Infect. Dis. 2006, 12, 569–574. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, L.; Compans, R.W.; Wang, B.Z. Universal influenza vaccines, a dream to be realized soon. Viruses 2014, 6, 1974–1991. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.T.; Kim, K.H.; Ko, E.J.; Lee, Y.N.; Kim, M.C.; Kwon, Y.M.; Tang, Y.; Cho, M.K.; Lee, Y.J.; Kang, S.M. New vaccines against influenza virus. Clin. Exp. Vaccine Res. 2014, 3, 12–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaiswal, V.; Chanumolu, S.K.; Sharma, P.; Chauhan, R.S.; Rout, C. EpiCombFlu: Exploring known influenza epitopes and their combination to design a universal influenza vaccine. Bioinformatics 2013, 29, 1904–1907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moise, L.; Terry, F.; Ardito, M.; Tassone, R.; Latimer, H.; Boyle, C.; Martin, W.D.; De Groot, A.S. Universal H1N1 influenza vaccine development: Identification of consensus class II hemagglutinin and neuraminidase epitopes derived from strains circulating between 1980 and 2011. Hum. Vaccines Immunother. 2013, 9, 1598–1607. [Google Scholar] [CrossRef] [Green Version]
- Nachbagauer, R.; Krammer, F. Universal influenza virus vaccines and therapeutic antibodies. Clin. Microbiol. Infect. 2017, 23, 222–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altenburg, A.F.; Kreijtz, J.H.; de Vries, R.D.; Song, F.; Fux, R.; Rimmelzwaan, G.F.; Sutter, G.; Volz, A. Modified vaccinia virus ankara (MVA) as production platform for vaccines against influenza and other viral respiratory diseases. Viruses 2014, 6, 2735–2761. [Google Scholar] [CrossRef]
- Volz, A.; Sutter, G. Modified vaccinia virus ankara: History, Value in basic research, and current perspectives for vaccine development. Adv. Virus Res. 2017, 97, 187–243. [Google Scholar] [CrossRef] [PubMed]
- Ramezanpour, B.; Pronker, E.S.; Kreijtz, J.H.; Osterhaus, A.D.; Claassen, E. Market implementation of the MVA platform for pre-pandemic and pandemic influenza vaccines: A quantitative key opinion leader analysis. Vaccine 2015, 33, 4349–4358. [Google Scholar] [CrossRef] [Green Version]
- Lambe, T.; Carey, J.B.; Li, Y.; Spencer, A.J.; van Laarhoven, A.; Mullarkey, C.E.; Vrdoljak, A.; Moore, A.C.; Gilbert, S.C. Immunity against heterosubtypic influenza virus induced by adenovirus and MVA expressing nucleoprotein and matrix protein-1. Sci. Rep. 2013, 3, 1443. [Google Scholar] [CrossRef] [Green Version]
- Hessel, A.; Savidis-Dacho, H.; Coulibaly, S.; Portsmouth, D.; Kreil, T.R.; Crowe, B.A.; Schwendinger, M.G.; Pilz, A.; Barrett, P.N.; Falkner, F.G.; et al. MVA vectors expressing conserved influenza proteins protect mice against lethal challenge with H5N1, H9N2 and H7N1 viruses. PLoS ONE 2014, 9, e88340. [Google Scholar] [CrossRef]
- Brewoo, J.N.; Powell, T.D.; Jones, J.C.; Gundlach, N.A.; Young, G.R.; Chu, H.; Das, S.C.; Partidos, C.D.; Stinchcomb, D.T.; Osorio, J.E. Cross-protective immunity against multiple influenza virus subtypes by a novel modified vaccinia Ankara (MVA) vectored vaccine in mice. Vaccine 2013, 31, 1848–1855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antrobus, R.D.; Lillie, P.J.; Berthoud, T.K.; Spencer, A.J.; McLaren, J.E.; Ladell, K.; Lambe, T.; Milicic, A.; Price, D.A.; Hill, A.V.; et al. A T cell-inducing influenza vaccine for the elderly: Safety and immunogenicity of MVA-NP+M1 in adults aged over 50 years. PLoS ONE 2012, 7, e48322. [Google Scholar] [CrossRef]
- Lillie, P.J.; Berthoud, T.K.; Powell, T.J.; Lambe, T.; Mullarkey, C.; Spencer, A.J.; Hamill, M.; Peng, Y.; Blais, M.E.; Duncan, C.J.; et al. Preliminary assessment of the efficacy of a T-cell-based influenza vaccine, MVA-NP+M1, in humans. Clin. Infect. Dis. 2012, 55, 19–25. [Google Scholar] [CrossRef]
- Folegatti, P.M.; Bellamy, D.; Flaxman, A.; Mair, C.; Ellis, C.; Ramon, R.L.; Ramos Lopez, F.; Mitton, C.; Baker, M.; Poulton, I.; et al. Safety and immunogenicity of the heterosubtypic influenza a vaccine MVA-NP+M1 manufactured on the AGE1.CR.pIX avian cell line. Vaccines 2019, 7, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prabakaran, M.; Leyrer, S.; He, F.; Auer, S.; Kumar, S.R.; Kindsmueller, K.; Mytle, N.; Schneider, J.; Lockhart, S.; Kwang, J. Progress toward a universal H5N1 vaccine: A recombinant modified vaccinia virus Ankara-expressing trivalent hemagglutinin vaccine. PLoS ONE 2014, 9, e107316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, L.J.; Muench, H. A simple method of estimating fifty percent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Bodmer, H.C.; Pemberton, R.M.; Rothbard, J.B.; Askonas, B.A. Enhanced recognition of a modified peptide antigen by cytotoxic T cells specific for influenza nucleoprotein. Cell 1988, 52, 253–258. [Google Scholar] [CrossRef]
- Taylor, P.M.; Davey, J.; Howland, K.; Rothbard, J.B.; Askonas, B.A. Class I MHC molecules rather than other mouse genes dictate influenza epitope recognition by cytotoxic T cells. Immunogenetics 1987, 26, 267–272. [Google Scholar] [CrossRef]
- Gao, X.M.; Liew, F.Y.; Tite, J.P. Identification and characterization of T helper epitopes in the nucleoprotein of influenza A virus. J. Immunol. 1989, 143, 3007–3014. [Google Scholar]
- Rammensee, H.G.; Friede, T.; Stevanoviic, S. MHC ligands and peptide motifs: First listing. Immunogenetics 1995, 41, 178–228. [Google Scholar] [CrossRef]
- Tscharke, D.C.; Woo, W.P.; Sakala, I.G.; Sidney, J.; Sette, A.; Moss, D.J.; Bennink, J.R.; Karupiah, G.; Yewdell, J.W. Poxvirus CD8+ T-cell determinants and cross-reactivity in BALB/c mice. J. Virol. 2006, 80, 6318–6323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitelli, A.; Quirion, M.R.; Lo, C.Y.; Misplon, J.A.; Grabowska, A.K.; Pierantoni, A.; Ammendola, V.; Price, G.E.; Soboleski, M.R.; Cortese, R.; et al. Vaccination to conserved influenza antigens in mice using a novel Simian adenovirus vector, PanAd3, derived from the bonobo Pan paniscus. PLoS ONE 2013, 8, e55435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, L.Y.; Ha do, L.A.; Simmons, C.; de Jong, M.D.; Chau, N.V.; Schumacher, R.; Peng, Y.C.; McMichael, A.J.; Farrar, J.J.; Smith, G.L.; et al. Memory T cells established by seasonal human influenza A infection cross-react with avian influenza A (H5N1) in healthy individuals. J. Clin. Investig. 2008, 118, 3478–3490. [Google Scholar] [CrossRef]
- Liu, X.; Guo, J.; Han, S.; Yao, L.; Chen, A.; Yang, Q.; Bo, H.; Xu, P.; Yin, J.; Zhang, Z. Enhanced immune response induced by a potential influenza A vaccine based on branched M2e polypeptides linked to tuftsin. Vaccine 2012, 30, 6527–6533. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.C.; Feng, H.; Ahmed, T.; Compans, R.W.; Wang, B.Z. Virus-like particles containing the tetrameric ectodomain of influenza matrix protein 2 and flagellin induce heterosubtypic protection in mice. BioMed Res. Int. 2013, 2013, 686549. [Google Scholar] [CrossRef] [Green Version]
- Deng, L.; Kim, J.R.; Chang, T.Z.; Zhang, H.; Mohan, T.; Champion, J.A.; Wang, B.Z. Protein nanoparticle vaccine based on flagellin carrier fused to influenza conserved epitopes confers full protection against influenza a virus challenge. Virology 2017, 509, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.C.; Lee, J.S.; Kwon, Y.M.; Eunju, O.; Lee, Y.J.; Choi, J.G.; Wang, B.Z.; Compans, R.W.; Kang, S.M. Multiple heterologous M2 extracellular domains presented on virus-like particles confer broader and stronger M2 immunity than live influenza a virus infection. Antivir. Res. 2013, 99, 328–335. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Hess, A.; Chang, T.Z.; Wang, Y.C.; Champion, J.A.; Compans, R.W.; Wang, B.Z. Nanoclusters self-assembled from conformation-stabilized influenza M2e as broadly cross-protective influenza vaccines. Nanomedicine 2014, 10, 473–482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Huang, B.; Jiang, T.; Wang, X.; Qi, X.; Gao, Y.; Tan, W.; Ruan, L. Robust immunity and heterologous protection against influenza in mice elicited by a novel recombinant NP-M2e fusion protein expressed in E. coli. PLoS ONE 2012, 7, e52488. [Google Scholar] [CrossRef]
- Luo, M.; Tao, P.; Li, J.; Zhou, S.; Guo, D.; Pan, Z. Immunization with plasmid DNA encoding influenza A virus nucleoprotein fused to a tissue plasminogen activator signal sequence elicits strong immune responses and protection against H5N1 challenge in mice. J. Virol. Methods 2008, 154, 121–127. [Google Scholar] [CrossRef]
- Roy, S.; Kobinger, G.P.; Lin, J.; Figueredo, J.; Calcedo, R.; Kobasa, D.; Wilson, J.M. Partial protection against H5N1 influenza in mice with a single dose of a chimpanzee adenovirus vector expressing nucleoprotein. Vaccine 2007, 25, 6845–6851. [Google Scholar] [CrossRef]
- Huang, B.; Wang, W.; Li, R.; Wang, X.; Jiang, T.; Qi, X.; Gao, Y.; Tan, W.; Ruan, L. Influenza a virus nucleoprotein derived from Escherichia coli or recombinant vaccinia (Tiantan) virus elicits robust cross-protection in mice. Virol. J. 2012, 9, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Gabbard, J.D.; Mooney, A.; Gao, X.; Chen, Z.; Place, R.J.; Tompkins, S.M.; He, B. Single-dose vaccination of a recombinant parainfluenza virus 5 expressing NP from H5N1 virus provides broad immunity against influenza A viruses. J. Virol. 2013, 87, 5985–5993. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H.; Kim, J.Y.; Choi, Y.; Nguyen, H.H.; Song, M.K.; Chang, J. Mucosal vaccination with recombinant adenovirus encoding nucleoprotein provides potent protection against influenza virus infection. PLoS ONE 2013, 8, e75460. [Google Scholar] [CrossRef] [Green Version]
- Epstein, S.L.; Kong, W.P.; Misplon, J.A.; Lo, C.Y.; Tumpey, T.M.; Xu, L.; Nabel, G.J. Protection against multiple influenza A subtypes by vaccination with highly conserved nucleoprotein. Vaccine 2005, 23, 5404–5410. [Google Scholar] [CrossRef]
- Lawson, C.M.; Bennink, J.R.; Restifo, N.P.; Yewdell, J.W.; Murphy, B.R. Primary pulmonary cytotoxic T lymphocytes induced by immunization with a vaccinia virus recombinant expressing influenza A virus nucleoprotein peptide do not protect mice against challenge. J. Virol. 1994, 68, 3505–3511. [Google Scholar] [CrossRef] [Green Version]
- LaMere, M.W.; Lam, H.T.; Moquin, A.; Haynes, L.; Lund, F.E.; Randall, T.D.; Kaminski, D.A. Contributions of antinucleoprotein IgG to heterosubtypic immunity against influenza virus. J. Immunol. 2011, 186, 4331–4339. [Google Scholar] [CrossRef] [Green Version]
- Leon, B.; Ballesteros-Tato, A.; Randall, T.D.; Lund, F.E. Prolonged antigen presentation by immune complex-binding dendritic cells programs the proliferative capacity of memory CD8 T cells. J. Exp. Med. 2014, 211, 1637–1655. [Google Scholar] [CrossRef] [Green Version]
- Levi, R.; Aboud-Pirak, E.; Leclerc, C.; Lowell, G.H.; Arnon, R. Intranasal immunization of mice against influenza with synthetic peptides anchored to proteosomes. Vaccine 1995, 13, 1353–1359. [Google Scholar] [CrossRef]
- Freer, G.; Senesi, S. No recognition of MHC class II+ cells infected with a vaccinia virus encoding influenza type A nucleoprotein by class II-restricted T cells. Immunol. Lett. 1993, 36, 305–312. [Google Scholar] [CrossRef]
- Beignon, A.S.; Briand, J.P.; Muller, S.; Partidos, C.D. Immunization onto bare skin with synthetic peptides: Immunomodulation with a CpG-containing oligodeoxynucleotide and effective priming of influenza virus-specific CD4+ T cells. Immunology 2002, 105, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Gui, X.; Ge, P.; Wang, X.; Yang, K.; Yu, H.; Zhao, Q.; Chen, Y.; Xia, N. Identification of a highly conserved and surface exposed B-cell epitope on the nucleoprotein of influenza A virus. J. Med. Virol. 2014, 86, 995–1002. [Google Scholar] [CrossRef]
- Yang, M.; Berhane, Y.; Salo, T.; Li, M.; Hole, K.; Clavijo, A. Development and application of monoclonal antibodies against avian influenza virus nucleoprotein. J. Virol. Methods 2008, 147, 265–274. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Zou, P.; Chen, Y.H. Monoclonal antibodies recognizing EVETPIRN epitope of influenza A virus M2 protein could protect mice from lethal influenza A virus challenge. Immunol. Lett. 2004, 93, 131–136. [Google Scholar] [CrossRef] [PubMed]
- Zou, P.; Liu, W.; Wu, F.; Chen, Y.H. Fine-epitope mapping of an antibody that binds the ectodomain of influenza matrix protein 2. FEMS Immunol. Med. Microbiol. 2008, 53, 79–84. [Google Scholar] [CrossRef] [PubMed]
- Huleatt, J.W.; Nakaar, V.; Desai, P.; Huang, Y.; Hewitt, D.; Jacobs, A.; Tang, J.; McDonald, W.; Song, L.; Evans, R.K.; et al. Potent immunogenicity and efficacy of a universal influenza vaccine candidate comprising a recombinant fusion protein linking influenza M2e to the TLR5 ligand flagellin. Vaccine 2008, 26, 201–214. [Google Scholar] [CrossRef]
- Ito, T.; Gorman, O.T.; Kawaoka, Y.; Bean, W.J.; Webster, R.G. Evolutionary analysis of the influenza A virus M gene with comparison of the M1 and M2 proteins. J. Virol. 1991, 65, 5491–5498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinder, D.; Bedford, T.; Gupta, S.; Pascual, M. The roles of competition and mutation in shaping antigenic and genetic diversity in influenza. PLoS Pathog. 2013, 9, e1003104. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MVA-Encoded Vaccine Antigens | Challenge | ||
|---|---|---|---|
| PR8 H1N1 | pH1N1 | H5N1 | |
| MVAtor | 0/8 (0%) | 1/8 (13%) | 0/8 (0%) |
| PBS | 0/8 (0%) | 2/8 (25%) | |
| NP | 8/8 (100%) | ||
| M2 | 8/8 (100%) | ||
| M2TML | 8/8 (100%) | ||
| METRC | 8/8 (100%) | ||
| METRS | 8/8 (100%) | ||
| NP + M2 | 8/8 (100%) | ||
| NP + M2TML Bl 1 | 8/8 (100%) | ||
| NP + METRC Bl 1 | 8/8 (100%) | ||
| NP + METRS Bl 1 | 8/8 (100%) | ||
| M1 | 3/8 (38%) | ||
| NP + M1 Bl 1 | 7/8 (88%) | ||
| NP + M2 Bl 1 | 8/8 (100%) | ||
| NP + M2 + M1 Bl 1 | 8/8 (100%) | ||
| NP + METRC + M1 Bl 1 | 8/8 (100%) | 7/8 (88%) | |
| NP + METRC + M1 SI 2 | 4/8 (50%) | ||
| H5 (VN) HA | 8/8 (100%) | ||
| MVAtor | 0/8 (0%) | ||
| PR8 Prime, NP + METRC + M1 Bl 1 | 8/8 (100%) | ||
| PR8 Prime, NP + METRC + M1 SI 2 | 8/8 (100%) | ||
| PR8 Prime, MVAtor | 8/8 (100%) | ||
| Immunization or Challenge → | 1 | 2 | 3 |
|---|---|---|---|
| Experiment One: two administrations of sublethal INFV A followed by sublethal INFV A or lethal INFV A | Sublethal Influenza N = 25 mice HAI titer = 436 ± 141 | Sublethal Influenza N = 25 mice HAI titer = 1173 ± 478 | Sublethal Influenza N = 5 mice HAI titer = 1440 ± 556 |
| Lethal Challenge N = 16 HAI titer = 1307 ± 596 | |||
| Experiment Two: two administrations of MVA HA vaccine, followed by MVA HA or lethal INFV A | MVA HA N = 25 mice HAI titer = 128 ± 535 | MVA HA N = 25 mice HAI titer = 1182 ± 653 | MVA HA N = 5 mice HAI titer = 981 ± 324 |
| Lethal Challenge N = 16 HAI titer = 953 ± 458 |
| Statistical Difference Compared to Medium | Recall Antigen | Antigen | MHC Class | Spots Per 300,000 Cells |
|---|---|---|---|---|
| Medium | None | None | 3 ± 2 | |
| p < 0.001 | NP (H1N1) protein | NP | NA 1 | 98 ± 56 |
| p < 0.001 | TYQRTRALV peptide | NP | I | 325 ± 126 |
| p < 0.001 | RLIQNSLTIERMVLS peptide | NP | II | 53 ± 31 |
| None | M2 protein (H1N1 PR8) | M2 | NA 1 | 11 ± 17 |
| None | IAANIIGIL peptide 2 | M2 | I | 7 ± 11 |
| None | IYRRFKYGL peptide 2 | M2 | I | 3 ± 2 |
| None | LLTEVETPI peptide | M2e | I | 7 ± 11 |
| p < 0.001 | VETPIRNEW peptide | M2e | I | 38 ± 18 |
| p < 0.001 | SLLTEVETPIRNEWGCRCNGSSD peptide | M2e | II | 26 ± 18 |
| NA 3 | SPGAAGWDL and VGPSNSPTF | MVA | I | 666 ± 71 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mytle, N.; Leyrer, S.; Inglefield, J.R.; Harris, A.M.; Hickey, T.E.; Minang, J.; Lu, H.; Ma, Z.; Andersen, H.; Grubaugh, N.D.; et al. Influenza Antigens NP and M2 Confer Cross Protection to BALB/c Mice against Lethal Challenge with H1N1, Pandemic H1N1 or H5N1 Influenza A Viruses. Viruses 2021, 13, 1708. https://doi.org/10.3390/v13091708
Mytle N, Leyrer S, Inglefield JR, Harris AM, Hickey TE, Minang J, Lu H, Ma Z, Andersen H, Grubaugh ND, et al. Influenza Antigens NP and M2 Confer Cross Protection to BALB/c Mice against Lethal Challenge with H1N1, Pandemic H1N1 or H5N1 Influenza A Viruses. Viruses. 2021; 13(9):1708. https://doi.org/10.3390/v13091708
Chicago/Turabian StyleMytle, Nutan, Sonja Leyrer, Jon R. Inglefield, Andrea M. Harris, Thomas E. Hickey, Jacob Minang, Hang Lu, Zhidong Ma, Hanné Andersen, Nathan D. Grubaugh, and et al. 2021. "Influenza Antigens NP and M2 Confer Cross Protection to BALB/c Mice against Lethal Challenge with H1N1, Pandemic H1N1 or H5N1 Influenza A Viruses" Viruses 13, no. 9: 1708. https://doi.org/10.3390/v13091708
APA StyleMytle, N., Leyrer, S., Inglefield, J. R., Harris, A. M., Hickey, T. E., Minang, J., Lu, H., Ma, Z., Andersen, H., Grubaugh, N. D., Guina, T., Skiadopoulos, M. H., & Lacy, M. J. (2021). Influenza Antigens NP and M2 Confer Cross Protection to BALB/c Mice against Lethal Challenge with H1N1, Pandemic H1N1 or H5N1 Influenza A Viruses. Viruses, 13(9), 1708. https://doi.org/10.3390/v13091708