
Generation of Nucleic Acid Aptamer Candidates against a Novel Calicivirus Protein Target

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cloning and Expression of VPgs
2.2. Aptamer Selection for Human Norovirus and Tulane Virus VPgs
2.2.1. Preparation of the ssDNA Library
2.2.2. Preparation of GST-VPg Protein Target
2.2.3. SELEX and Counter-SELEX Process
2.2.4. Analysis of Aptamer Sequences, Structural Folding, and Stability
2.2.5. Overexpression of VPg Proteins
2.2.6. Preparation of Crude Extracts
2.2.7. Enzyme-Linked Aptamer Sorbent Assay (ELASA)
2.2.8. ELASA Optimization and Validation
2.2.9. Binding Affinity Analysis
3. Results
3.1. Aptamer Candidates
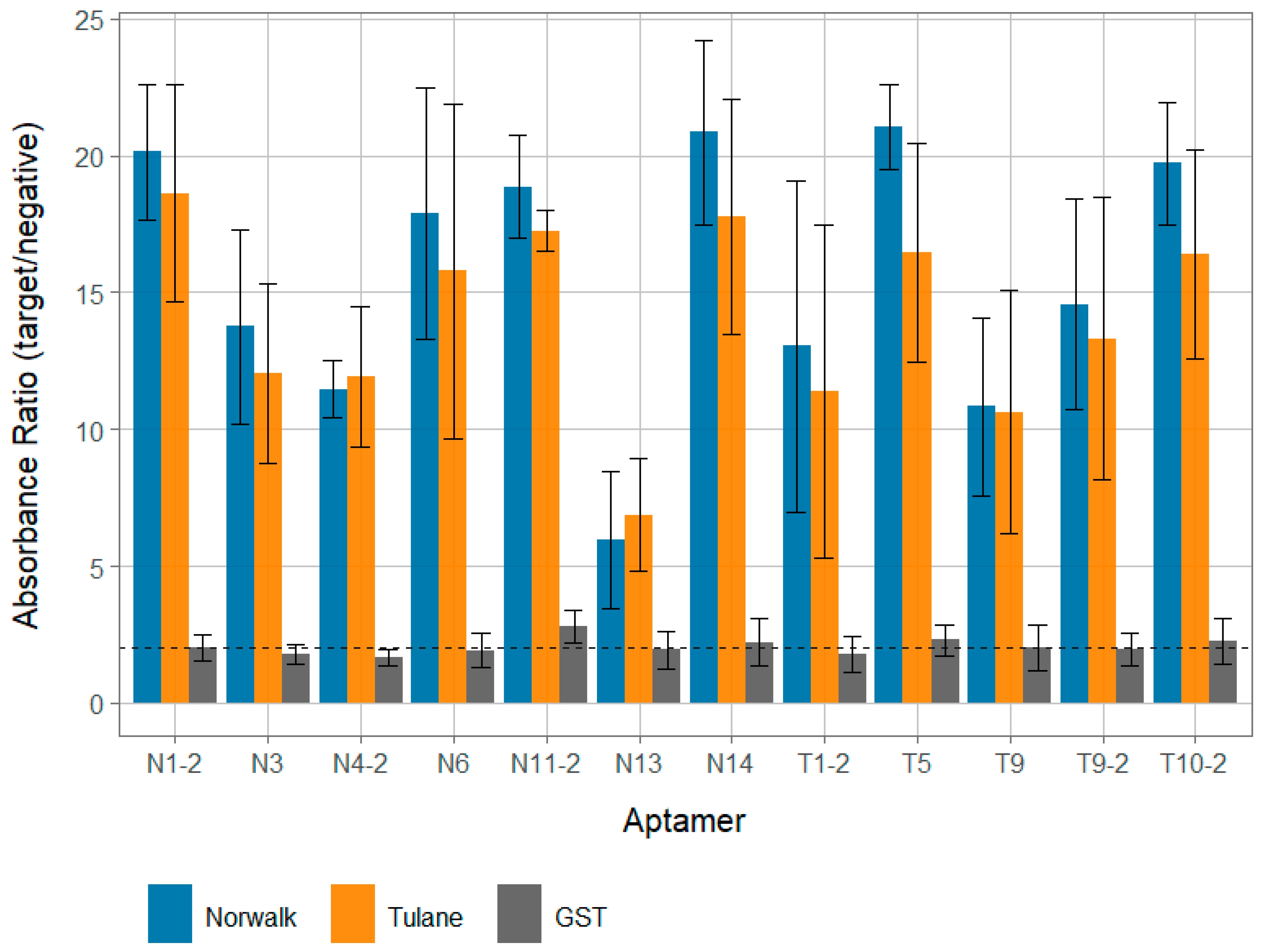
3.2. Aptamer Reactivty
3.3. Aptamer Specificity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Kirk, M.D.; Pires, S.M.; Black, R.E.; Caipo, M.; Crump, J.A.; Devleesschauwer, B.; Dopfer, D.; Fazil, A.; FIscher-Walker, C.L.; Hald, T.; et al. World Health Organization Estimates of the Global and Regional Disease Burden of 11 Foodborne Bacterial, Protozoal, and Viral Diseases, 2010: A Data Synthesis. PLoS Med. 2015, 12, e1001920. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.D.; Goulter, R.M.; Jaykus, L.-A. Human Norovirus as a Foodborne Pathogen: Challenges and Developments. Annu. Rev. Food Sci. Technol. 2015, 6, 411–433. [Google Scholar] [CrossRef]
- Vinjé, J. Advances in Laboratory Methods for Detection and Typing of Norovirus. J. Clin. Microbiol. 2015, 53, 373–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiota, T.; Okame, M.; Takanashi, S.; Khamrin, P.; Takagi, M.; Satou, K.; Masuoka, Y.; Yagyu, F.; Shimizu, Y.; Kohno, H.; et al. Characterization of a broadly reactive monoclonal antibody against norovirus genogroups I and II: Recognition of a novel conformational epitope. J. Virol. 2007, 81, 12298–12306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, M.D.; Escudero-Abarca, B.I.; Suh, S.H.; Jaykus, L.-A. Generation and Characterization of Nucleic Acid Aptamers Targeting the Capsid P Domain of a Human Norovirus GII.4 Strain. J. Biotechnol. 2015, 209, 41–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, T.D.; Kitamoto, N.; Hutson, A.M.; Estes, M.K.; Tanaka, T. Identification of Genogroup I and Genogroup II Broadly Reactive Epitopes on the Norovirus Capsid. J. Virol. 2005, 79, 7402–7409. [Google Scholar] [CrossRef] [Green Version]
- Manuel, C.S.; Moore, M.D.; Jaykus, L.-A. Predicting human norovirus infectivity: Recent advances and continued challenges. Food Microbiol. 2018, 76, 337–345. [Google Scholar] [CrossRef]
- Li, X.; Huang, R.; Chen, H. Evaluation of Assays to Quantify Infectious Human Norovirus for Heat and High-Pressure Inactivation Studies Using Tulane Virus. Food Environ. Virol. 2017, 9, 314–325. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Lindesmith, L.C.; Lobue, A.D.; Baric, R.S. Viral shape-shifting: Norovirus evasion of the human immune system. Nat. Rev. Microbiol. 2010, 8, 231–241. [Google Scholar] [CrossRef]
- Shirato, H. Norovirus and histo-blood group antigens. Jpn. J. Infect. Dis. 2011, 64, 95–103. [Google Scholar]
- Murakami, K.; Kurihara, C.; Oka, T.; Shimoike, T.; Fujii, Y.; Takai-Todaka, R.; Park, Y.; Wakita, T.; Matsuda, T.; Hokari, R.; et al. Norovirus binding to intestinal epithelial cells is independent of histo-blood group antigens. PLoS ONE 2013, 8, e66534. [Google Scholar] [CrossRef] [Green Version]
- Huang, P.; Farkas, T.; Zhong, W.; Tan, M.; Thornton, S.; Morrow, A.L.; Jiang, X.; Harbor, P. Norovirus and Histo-Blood Group Antigens: Demonstration of a Wide Spectrum of Strain Specificities and Classification of Two Major Binding Groups among Multiple Binding Patterns. J. Virol. 2005, 79, 6714–6722. [Google Scholar] [CrossRef] [Green Version]
- Harrington, P.R.; Lindesmith, L.; Yount, B.; Moe, C.L.; Baric, R.S. Binding of Norwalk Virus-Like Particles to ABH Histo-Blood Group Antigens Is Blocked by Antisera from Infected Human Volunteers or Experimentally Vaccinated Mice. J. Virol. 2002, 76, 12335–12343. [Google Scholar] [CrossRef] [Green Version]
- Harrington, P.R.; Vinje, J.; Moe, C.L.; Baric, R.S. Norovirus Capture with Histo-Blood Group Antigens Reveals Novel Virus-Ligand Interactions. J. Virol. 2004, 78, 3035–3045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bunka, D.H.J.; Platonova, O.; Stockley, P.G. Development of aptamer therapeutics. Curr. Opin. Pharmacol. 2010, 10, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Binning, J.M.; Leung, D.W.; Amarasinghe, G.K. Aptamers in virology: Recent advances and challenges. Front. Microbiol. 2012, 3, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tombelli, S.; Minunni, M.; Mascini, M. Aptamers-based assays for diagnostics, environmental and food analysis. Biomol. Eng. 2007, 24, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Escudero-Abarca, B.I.; Suh, S.H.; Moore, M.D.; Dwivedi, H.P.; Jaykus, L.-A. Selection, Characterization and Application of Nucleic Acid Aptamers for the Capture and Detection of Human Norovirus Strains. PLoS ONE 2014, 9, e106805. [Google Scholar] [CrossRef] [PubMed]
- Giamberardino, A.; Labib, M.; Hassan, E.M.; Tetro, J.A.; Springthorpe, S.; Sattar, S.A.; Berezovski, M.V.; Derosa, M.C. Ultrasensitive norovirus detection using DNA aptasensor technology. PLoS ONE 2013, 8, e79087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beier, R.; Pahlke, C.; Quenzel, P.; Henseleit, A.; Boschke, E.; Cuniberti, G.; Labudde, D. Selection of a DNA aptamer against norovirus capsid protein VP1. FEMS Microbiol. Lett. 2014, 351, 162–169. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.D.; Bobay, B.G.; Mertens, B.; Jaykus, L. Human norovirus aptamer exhibits high degree of target conformation-dependent binding similar to that of receptors and discriminates particle functionality. mSphere 2016, 1, e00298-16. [Google Scholar] [CrossRef] [Green Version]
- Goodfellow, I. The genome-linked protein VPg of vertebrate viruses—A multifaceted protein. Curr. Opin. Virol. 2011, 1, 355–362. [Google Scholar] [CrossRef] [Green Version]
- Thorne, L.G.; Goodfellow, I.G. Norovirus gene expression and replication. J. Gen. Virol. 2014, 95, 278–291. [Google Scholar] [CrossRef]
- Leen, E.N.; Sorgeloos, F.; Correia, S.; Chaudhry, Y.; Cannac, F.; Pastore, C.; Xu, Y.; Graham, S.C.; Matthews, S.J.; Goodfellow, I.G.; et al. A Conserved Interaction between a C-Terminal Motif in Norovirus VPg and the HEAT-1 Domain of eIF4G Is Essential for Translation Initiation. PLoS Pathog. 2016, 12, 1–34. [Google Scholar] [CrossRef] [Green Version]
- Guix, S.; Asanaka, M.; Katayama, K.; Crawford, S.E.; Neill, F.H.; Atmar, R.L.; Estes, M.K. Norwalk virus RNA is infectious in mammalian cells. J. Virol. 2007, 81, 12238–12248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farkas, T.; Sestak, K.; Wei, C.; Jiang, X. Characterization of a rhesus monkey calicivirus representing a new genus of Caliciviridae. J. Virol. 2008, 82, 5408–5416. [Google Scholar] [CrossRef] [Green Version]
- Moorman, E.; Montazeri, N.; Jaykus, L. Efficacy of neutral electrolyzed water for inactivation of human norovirus. Appl. Environ. Microbiol. 2017, 83, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daughenbaugh, K.F.; Fraser, C.S.; Hershey, J.W.B.; Hardy, M.E. The genome-linked protein VPg of the Norwalk virus binds eIF3, suggesting its role in translation initiation complex recruitment. EMBO J. 2003, 22, 2852–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dwivedi, H.P.; Smiley, R.D.; Jaykus, L.-A. Selection and characterization of DNA aptamers with binding selectivity to Campylobacter jejuni using whole-cell SELEX. Appl. Microbiol. Biotechnol. 2010, 87, 2323–2334. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. In Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology, Stanford, CA, USA, 14–17 August 1994; Volume 2, pp. 28–36. [Google Scholar]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef]
- Moore, M.D.; Escudero-Abarca, B.I.; Jaykus, L.-A. An enzyme-linked aptamer sorbent assay to evaluate aptamer binding. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2017; Volume 1575, pp. 291–302. ISBN 978-1-4939-6855-8. [Google Scholar]
- Ebel, G.D.; Dupuis, A.P.; Nicholas, D.; Young, D.; Maffei, J.; Kramer, L.D. Detection by enzyme-linked immunosorbent assay of antibodies to West Nile virus in birds. Emerg. Infect. Dis. 2002, 8, 979–982. [Google Scholar] [CrossRef]
- Hirneisen, K.A.; Kniel, K.E. Comparison of ELISA attachment and infectivity assays for murine norovirus. J. Virol. Methods 2012, 186, 14–20. [Google Scholar] [CrossRef]
- Daughenbaugh, K.F.; Wobus, C.E.; Hardy, M.E. VPg of murine norovirus binds translation initiation factors in infected cells. Virol. J. 2006, 3, 33. [Google Scholar] [CrossRef] [Green Version]
- Leen, E.N.; Kwok, K.Y.R.; Birtley, J.R.; Simpson, P.J.; Subba-Reddy, C.V.; Chaudhry, Y.; Sosnovtsev, S.V.; Green, K.Y.; Prater, S.N.; Tong, M.; et al. Structures of the Compact Helical Core Domains of Feline Calicivirus and Murine Norovirus VPg Proteins. J. Virol. 2013, 87, 5318–5330. [Google Scholar] [CrossRef] [Green Version]
- Knight, A.; Li, D.; Uyttendaele, M.; Jaykus, L.-A. A critical review of methods for detecting human noroviruses and predicting their infectivity. Crit. Rev. Microbiol. 2013, 39, 295–309. [Google Scholar] [CrossRef]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.-L.; Qu, L.; et al. Replication of human noroviruses in stem cell–derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.K.; Watanabe, M.; Zhu, S.; Graves, C.L.; Keyes, L.R.; Grau, K.R.; Gonzalez-Hernandez, M.B.; Iovine, N.M.; Wobus, C.E.; Vinje, J.; et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science 2014, 346, 755–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rocha-Pereira, J.; Neyts, J.; Jochmans, D. Norovirus: Targets and tools in antiviral drug discovery. Biochem. Pharmacol. 2014, 91, 1–11. [Google Scholar] [CrossRef]
- Subba-Reddy, C.V.; Goodfellow, I.; Kao, C.C. VPg-primed RNA synthesis of norovirus RNA-dependent RNA polymerases by using a novel cell-based assay. J. Virol. 2011, 85, 13027–13037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McSweeney, A.; Davies, C.; Ward, V.; McSweeney, A.; Davies, C.; Ward, V.K. Cell Cycle Arrest is a Conserved Function of Norovirus VPg Proteins. Viruses 2019, 11, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keefe, A.D.; Pai, S.; Ellington, A. Aptamers as therapeutics. Nat. Rev. Drug Discov. 2010, 9, 537–550. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Primer Name | Sequence (5′ to 3′) a | Reference |
|---|---|---|
| Tulane VPg Forward | CCGGAATTCGCCAAGGGCAAGACAAAAAGG | This work |
| Tulane VPg Reverse | CCGCTCGAGCTACTCGTCGTAATAATCATCACTGGG | This work |
| Norwalk VPg Forward | CCGGAATTCGGAAAGAACAAAGGCAAGACC | [28] |
| Norwalk VPg Reverse | CCGCTCGAGTTCAAAATTGATCTTTTCATTATAAT | [28] |
| DNA Aptamer Library | AGTATACGTATTACCTGCAGC-N40-CGATATCTCGGAGATCTTGC | [18,29] |
| Aptamer Forward Constant | AGTATACGTATTACCTGCAGC | [18,29] |
| Aptamer Reverse Constant | /Biotin/GCAAGATCTCCGAGATATCG | [18,29] |
| Name | Conserved Sequence a | G (kcal/mol) | Frequency |
|---|---|---|---|
| T5 | TCACACTCGTTTCTATTACTAAAACATCGTTCCTTTCAGC | −5.95 | 13/17 |
| T9 | TGGAAGGCGGGAAGATTTTTGGTCGACCTGACAACCCGGT | −10.19 | 1/17 |
| T1-2 | TAGTAACGATTACCAAAATTCTCCCGAGGCTGACAACCCG | −6.47 | 1/17 |
| T9-2 | TCGAGGTATGGCCTTGTCTAGGCGCACCTGACAACCCGGTG | −11.89 | 1/17 |
| T10-2 | TGTCGTTAATTATTCGTGATCTGACAACCCGATCACTCTC | −12.01 | 1/17 |
| Name | Conserved Sequence a | G (kcal/mol) | Frequency |
|---|---|---|---|
| N3 | AGGGATGTGTTGGATGCATGCCAGGCTTGGTAACATTGTA | −9.90 | 1/19 |
| N6 | CAGAGTTGATGTAAGCTTCGTGTTAGCTCAACTCTTATCG | −8.36 | 9/19 |
| N13 | TCTTCGGTTTAATAAAGTTGGCTAGGAAAGTTTAAAACCG | −7.04 | 3/19 |
| N14 | AGTGGGTGGTGATGAATTCTGGTCGCGCTGACAACCCGCG | −11.90 | 1/19 |
| N1-2 | CGGGTCTCGTCTATGCAGTACTCAAAACGCTTGAGGTACCGA | −11.92 | 1/19 |
| N3-2 b | CAGAGTTGATGTAAGCTTCGTGTTAGCTTAACTCTTATCG | −7.87 | 1/19 |
| N4-2 | AAGGCTTTTTTAAAGGCTAGGCTTGATAATCGGTTAACTC | −13.46 | 1/19 |
| N11-2 | TGTCGATAAAGTGAGTTAAGTCACCGGCCCGGCCTATTCG | −6.63 | 1/19 |
| N12-2 c | TCACACTCGTTTCTATTACTAAAACATCGTTCCTTTCAGC | −5.95 | 1/19 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Faircloth, J.; Moore, M.D.; Stoufer, S.; Kim, M.; Jaykus, L.-A. Generation of Nucleic Acid Aptamer Candidates against a Novel Calicivirus Protein Target. Viruses 2021, 13, 1716. https://doi.org/10.3390/v13091716
Faircloth J, Moore MD, Stoufer S, Kim M, Jaykus L-A. Generation of Nucleic Acid Aptamer Candidates against a Novel Calicivirus Protein Target. Viruses. 2021; 13(9):1716. https://doi.org/10.3390/v13091716
Chicago/Turabian StyleFaircloth, Jeremy, Matthew D. Moore, Sloane Stoufer, Minji Kim, and Lee-Ann Jaykus. 2021. "Generation of Nucleic Acid Aptamer Candidates against a Novel Calicivirus Protein Target" Viruses 13, no. 9: 1716. https://doi.org/10.3390/v13091716
APA StyleFaircloth, J., Moore, M. D., Stoufer, S., Kim, M., & Jaykus, L. -A. (2021). Generation of Nucleic Acid Aptamer Candidates against a Novel Calicivirus Protein Target. Viruses, 13(9), 1716. https://doi.org/10.3390/v13091716