
Genotype of Immunologically Hot or Cold Tumors Determines the Antitumor Immune Response and Efficacy by Fully Virulent Retargeted oHSV


 , ,
, ,
Abstract
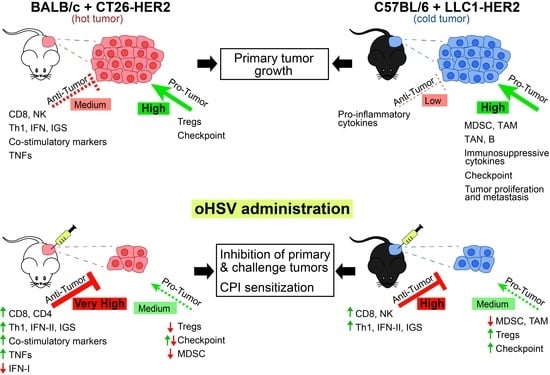
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Line and Virus
2.2. Mice and In Vivo Experiments
2.3. Characterization of Tumor Microenvironment Infiltrating Cells
2.4. Determination of Intratumoral Cytokine and Immune Factors by RT-PCR and Expression Arrays
2.5. Detection of Splenocyte Reactivity to Cancer Cells
2.6. Detection of Serum Antibodies to Cancer Cells and to HSV
3. Results
3.1. Generation of CT26-HER2 Cells and Their Engraftment in HER2 Transgenic/Tolerant BALB/c Mice
3.2. Immune Profiling of CT26-HER2 Tumors
3.3. Efficacy of R-337 Monotherapy against CT26-HER2 Primary Tumors
3.4. Efficacy of R-337 Monotherapy against CT26-HER2 Challenge Tumors
3.5. R-337-Induced Modifications to Tumor Microenvironment
3.6. Efficacy of R-337 and Anti-CTLA-4 Combination Treatment
4. Discussion
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Menotti, L.; Nicoletti, G.; Gatta, V.; Croci, S.; Landuzzi, L.; De Giovanni, C.; Nanni, P.; Lollini, P.L.; Campadelli-Fiume, G. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9039–9044. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Cerretani, A.; Hengel, H.; Campadelli-Fiume, G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J. Virol. 2008, 20, 10153–10161. [Google Scholar] [CrossRef] [Green Version]
- Nanni, P.; Nicoletti, G.; Landuzzi, L.; Croci, S.; Murgo, A.; Palladini, A.; Antognoli, A.; Ianzano, M.L.; Stivani, V.; Grosso, V.; et al. High metastatic efficiency of human sarcoma cells in Rag2/gammac double knockout mice provides a powerful test system for antimetastatic targeted therapy. Eur. J. Cancer 2010, 46, 659–668. [Google Scholar] [CrossRef]
- Campadelli-Fiume, G.; De Giovanni, C.; Gatta, V.; Nanni, P.; Lollini, P.L.; Menotti, L. Rethinking herpes simplex virus: The way to oncolytic agents. Rev. Med. Virol. 2011, 21, 213–226. [Google Scholar] [CrossRef]
- Leoni, V.; Gatta, V.; Casiraghi, C.; Nicosia, A.; Petrovic, B.; Campadelli-Fiume, G. A Strategy for Cultivation of Retargeted Oncolytic Herpes Simplex Viruses in Non-cancer Cells. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Leoni, V.; Vannini, A.; Gatta, V.; Rambaldi, J.; Sanapo, M.; Barboni, C.; Zaghini, A.; Nanni, P.; Lollini, P.L.; Casiraghi, C.; et al. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. 2018, 14, e1007209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrovic, B.; Gianni, T.; Gatta, V.; Campadelli-Fiume, G. Insertion of a ligand to HER2 in gB retargets HSV tropism and obviates the need for activation of the other entry glycoproteins. PLoS Pathog. 2017, 13, e1006352. [Google Scholar] [CrossRef]
- Gatta, V.; Petrovic, B.; Campadelli-Fiume, G. The engineering of a novel ligand in gH confers to HSV an expanded tropism independent of gD activation by its receptors. PLoS Pathog. 2015, 11, e1004907. [Google Scholar] [CrossRef] [Green Version]
- Menotti, L.; Avitabile, E. Herpes simplex virus oncolytic immunovirotherapy: The blossoming branch of multimodal therapy. Int. J. Mol. Sci. 2020, 21, 8310. [Google Scholar] [CrossRef]
- Cheever, M.A.; Allison, J.P.; Ferris, A.S.; Finn, O.J.; Hastings, B.M.; Hecht, T.T.; Mellman, I.; Prindiville, S.A.; Viner, J.L.; Weiner, L.M.; et al. The prioritization of cancer antigens: A national cancer institute pilot project for the acceleration of translational research. Clin. Cancer Res. 2009, 15, 5323–5337. [Google Scholar] [CrossRef] [Green Version]
- Slamon, D.J.; Clark, G.M.; Wong, S.G.; Levin, W.J.; Ullrich, A.; McGuire, W.L. Human breast cancer: Correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 1987, 235, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Chou, J.; Kern, E.R.; Whitley, R.J.; Roizman, B. Mapping of herpes simplex virus-1 neurovirulence to gamma 134.5, a gene nonessential for growth in culture. Science 1990, 250, 1262–1266. [Google Scholar] [CrossRef]
- Kangas, C.; Krawczyk, E.; He, B. Oncolytic HSV: Underpinnings of tumor susceptobility. Viruses 2021, 13, 1408. [Google Scholar] [CrossRef]
- Bernstock, J.D.; Hoffman, S.E.; Chen, J.A.; Gupta, S.; Kappel, A.D.; Smith, T.R.; Chiocca, E.A. The Current Landscape of Oncolytic Herpes Simplex Viruses as Novel Therapies for Brain Malignancies. Viruses 2021, 13, 1158. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.S.; Hecht, J.R.; Chan, E. Talimogene laherparepvec: Review of its mechanism of action and clinical efficacy and safety. Immunotherapy 2019, 11, 705–723. [Google Scholar] [CrossRef] [Green Version]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef] [PubMed]
- Andtbacka, R.H.; Collichio, F.; Harrington, K.J.; Middleton, M.R.; Downey, G.; Öhrling, K.; Kaufman, H.L. Final analyses of OPTiM: A randomized phase III trial of talimogene laherparepvec versus granulocyte-macrophage colony-stimulating factor in unresectable stage III–IV melanoma. J. Immunother. Cancer 2019, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, V.; Gatta, V.; Palladini, A.; Nicoletti, G.; Ranieri, D.; Dall’Ora, M.; Grosso, V.; Rossi, M.; Alviano, F.; Bonsi, L.; et al. Systemic delivery of HER2-retargeted oncolytic-HSV by mesenchymal stromal cells protects from lung and brain metastases. Oncotarget 2015, 6, 34774. [Google Scholar] [CrossRef] [Green Version]
- Gambini, E.; Reisoli, E.; Appolloni, I.; Gatta, V.; Campadelli-Fiume, G.; Menotti, L.; Malatesta, P. Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol. Ther. 2012, 20, 994–1001. [Google Scholar] [CrossRef] [Green Version]
- De Lucia, M.; Cotugno, G.; Bignone, V.; Garzia, I.; Nocchi, L.; Langone, F.; Petrovic, B.; Sasso, E.; Pepe, S.; Froechlich, G.; et al. Retargeted and multi-cytokine armed herpes virus is a potent cancer endovaccine for local and systemic anti-tumor treatment in combination with anti-PD1. Mol. Ther. Oncolytics 2020, 19, 253–264. [Google Scholar] [CrossRef]
- Nanni, P.; Gatta, V.; Menotti, L.; De Giovanni, C.; Ianzano, M.; Palladini, A.; Grosso, V.; Dall’ora, M.; Croci, S.; Nicoletti, G.; et al. Preclinical Therapy of Disseminated HER-2(+) Ovarian and Breast Carcinomas with a HER-2-Retargeted Oncolytic Herpesvirus. PLoS Pathog. 2013, 9, e1003155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.S.; Fletcher, J.A.; Linette, G.P.; Stec, J.; Clark, E.; Ayers, M.; Symmans, W.F.; Pusztai, L.; Bloom, K.J. The Her-2/neu gene and protein in breast cancer 2003: Biomarker and target of therapy. Oncologist 2003, 8, 307–325. [Google Scholar] [CrossRef] [PubMed]
- Hainsworth, J.D.; Meric-Bernstam, F.; Swanton, C.; Hurwitz, H.; Spigel, D.R.; Sweeney, C.; Burris, H.; Bose, R.; Yoo, B.; Stein, A. Targeted therapy for advanced solid tumors on the basis of molecular profiles: Results from MyPathway, an open-label, phase IIa multiple basket study. J. Clin. Oncol. 2018, 36, 536–544. [Google Scholar] [CrossRef]
- Baselga, J.; Perez, E.A.; Pienkowski, T.; Bell, R. Adjuvant trastuzumab: A milestone in the treatment of HER-2-positive early breast cancer. Oncologist 2006, 11 (Suppl. 1), 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vannini, A.; Leoni, V.; Sanapo, M.; Gianni, T.; Giordani, G.; Gatta, V.; Barboni, C.; Zaghini, A.; Campadelli-Fiume, G. Immunotherapeutic Efficacy of Retargeted oHSVs Designed for Propagation in an Ad Hoc Cell Line. Cancers 2021, 13, 266. [Google Scholar] [CrossRef]
- Petrovic, B.; Leoni, V.; Gatta, V.; Zaghini, A.; Vannini, A.; Campadelli-Fiume, G. Dual Ligand Insertion in gB and gD of Oncolytic Herpes Simplex Viruses for Retargeting to a Producer Vero Cell Line and to Cancer Cells. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [Green Version]
- Lechner, M.G.; Karimi, S.S.; Barry-Holson, K.; Angell, T.E.; Murphy, K.A.; Church, C.H.; Ohlfest, J.R.; Hu, P.; Epstein, A.L. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J. Immunother. 2013, 36, 477–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.W.; Bhattacharya, S.; Yanamandra, N.; Kilian, D.; Shi, H.; Yadavilli, S.; Katlinskaya, Y.; Kaczynski, H.; Conner, M.; Benson, W. Tumor-immune profiling of murine syngeneic tumor models as a framework to guide mechanistic studies and predict therapy response in distinct tumor microenvironments. PLoS ONE 2018, 13, e0206223. [Google Scholar] [CrossRef]
- Mosely, S.I.; Prime, J.E.; Sainson, R.C.; Koopmann, J.-O.; Wang, D.Y.; Greenawalt, D.M.; Ahdesmaki, M.J.; Leyland, R.; Mullins, S.; Pacelli, L. Rational selection of syngeneic preclinical tumor models for immunotherapeutic drug discovery. Cancer Immunol. Res. 2017, 5, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Romero, K.; Rodríguez, R.M.; Amedei, A.; Barceló-Coblijn, G.; Lopez, D.H. Immune Landscape in Tumor Microenvironment: Implications for Biomarker Development and Immunotherapy. Int. J. Mol. Sci. 2020, 21, 5521. [Google Scholar] [CrossRef]
- Zhong, W.; Myers, J.S.; Wang, F.; Wang, K.; Lucas, J.; Rosfjord, E.; Lucas, J.; Hooper, A.T.; Yang, S.; Lemon, L.A. Comparison of the molecular and cellular phenotypes of common mouse syngeneic models with human tumors. BMC Genom. 2020, 21, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Rooney, M.S.; Shukla, S.A.; Wu, C.J.; Getz, G.; Hacohen, N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015, 160, 48–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science 2015, 348, 56–61. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castle, J.C.; Loewer, M.; Boegel, S.; de Graaf, J.; Bender, C.; Tadmor, A.D.; Boisguerin, V.; Bukur, T.; Sorn, P.; Paret, C. Immunomic, genomic and transcriptomic characterization of CT26 colorectal carcinoma. BMC Genom. 2014, 15, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Cocchi, F.; Fusco, D.; Menotti, L.; Gianni, T.; Eisenberg, R.J.; Cohen, G.H.; Campadelli-Fiume, G. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc. Natl. Acad. Sci. USA 2004, 101, 7445–7450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, C. Genetics of natural resistance to herpesvirus infections in mice. Nature 1975, 258, 152–153. [Google Scholar] [CrossRef]
- Yang, M.; Lu, J.; Zhang, G.; Wang, Y.; He, M.; Xu, Q.; Xu, C.; Liu, H. CXCL13 shapes immunoactive tumor microenvironment and enhances the efficacy of PD-1 checkpoint blockade in high-grade serous ovarian cancer. J. Immunother. Cancer 2021, 9. [Google Scholar] [CrossRef] [PubMed]
- Korbecki, J.; Grochans, S.; Gutowska, I.; Barczak, K.; Baranowska-Bosiacka, I. CC chemokines in a tumor: A review of pro-cancer and anti-cancer properties of receptors CCR5, CCR6, CCR7, CCR8, CCR9, and CCR10 ligands. Int. J. Mol. Sci. 2020, 21, 7619. [Google Scholar] [CrossRef]
- Marshall, L.A.; Marubayashi, S.; Jorapur, A.; Jacobson, S.; Zibinsky, M.; Robles, O.; Hu, D.X.; Jackson, J.J.; Pookot, D.; Sanchez, J. Tumors establish resistance to immunotherapy by regulating Treg recruitment via CCR4. J. Immunother. Cancer 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [Green Version]
- Park, J.M.; Terabe, M.; van den Broeke, L.T.; Donaldson, D.D.; Berzofsky, J.A. Unmasking immunosurveillance against a syngeneic colon cancer by elimination of CD4+ NKT regulatory cells and IL-13. Int. J. Cancer 2005, 114, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Prendergast, G.C.; Smith, C.; Thomas, S.; Mandik-Nayak, L.; Laury-Kleintop, L.; Metz, R.; Muller, A.J. Indoleamine 2, 3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Alvarado, D.M.; Iticovici, M.; Kau, N.S.; Park, H.; Parikh, P.J.; Thotala, D.; Ciorba, M.A. Interferon-Induced IDO1 mediates radiation resistance and is a therapeutic target in colorectal cancer. Cancer Immunol. Res. 2020, 8, 451–464. [Google Scholar] [CrossRef] [Green Version]
- Moesta, A.K.; Cooke, K.; Piasecki, J.; Mitchell, P.; Rottman, J.B.; Fitzgerald, K.; Zhan, J.; Yang, B.; Le, T.; Belmontes, B.; et al. Local Delivery of OncoVEXmGM-CSF Generates Systemic Antitumor Immune Responses Enhanced by Cytotoxic T-Lymphocyte-Associated Protein Blockade. Clin. Cancer Res. 2017, 23, 6190–6202. [Google Scholar] [CrossRef] [Green Version]
- Vannini, A.; Leoni, V.; Campadelli-Fiume, G. Targeted Delivery of IL-12 adjuvants immunotherapy by oncolytic viruses. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2021; p. 1290. [Google Scholar]
- Smith, K.D.; Shao, M.Y.; Posner, M.C.; Weichselbaum, R.R. Tumor genotype determines susceptibility to oncolytic herpes simplex virus mutants: Strategies for clinical application. Future Med. 2007, 3, 545–556. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gianni, T.; Leoni, V.; Sanapo, M.; Parenti, F.; Bressanin, D.; Barboni, C.; Zaghini, A.; Campadelli-Fiume, G.; Vannini, A. Genotype of Immunologically Hot or Cold Tumors Determines the Antitumor Immune Response and Efficacy by Fully Virulent Retargeted oHSV. Viruses 2021, 13, 1747. https://doi.org/10.3390/v13091747
Gianni T, Leoni V, Sanapo M, Parenti F, Bressanin D, Barboni C, Zaghini A, Campadelli-Fiume G, Vannini A. Genotype of Immunologically Hot or Cold Tumors Determines the Antitumor Immune Response and Efficacy by Fully Virulent Retargeted oHSV. Viruses. 2021; 13(9):1747. https://doi.org/10.3390/v13091747
Chicago/Turabian StyleGianni, Tatiana, Valerio Leoni, Mara Sanapo, Federico Parenti, Daniela Bressanin, Catia Barboni, Anna Zaghini, Gabriella Campadelli-Fiume, and Andrea Vannini. 2021. "Genotype of Immunologically Hot or Cold Tumors Determines the Antitumor Immune Response and Efficacy by Fully Virulent Retargeted oHSV" Viruses 13, no. 9: 1747. https://doi.org/10.3390/v13091747
APA StyleGianni, T., Leoni, V., Sanapo, M., Parenti, F., Bressanin, D., Barboni, C., Zaghini, A., Campadelli-Fiume, G., & Vannini, A. (2021). Genotype of Immunologically Hot or Cold Tumors Determines the Antitumor Immune Response and Efficacy by Fully Virulent Retargeted oHSV. Viruses, 13(9), 1747. https://doi.org/10.3390/v13091747