
Metabolic Reprogramming of Glioblastoma Cells during HCMV Infection Induces Secretome-Mediated Paracrine Effects in the Microenvironment

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Cell and Virus Culture
2.2. xCELLigence Real-Time Cell Analysis
2.3. RNA Isolation and qRT-PCR
2.4. RNA-Seq
2.5. Seahorse Bioanalyzer
2.6. Western Blotting
2.7. Flow Cytometry
2.8. Lactate Assay
2.9. Co-Culture Assay
2.10. Statistical Analysis
3. Results
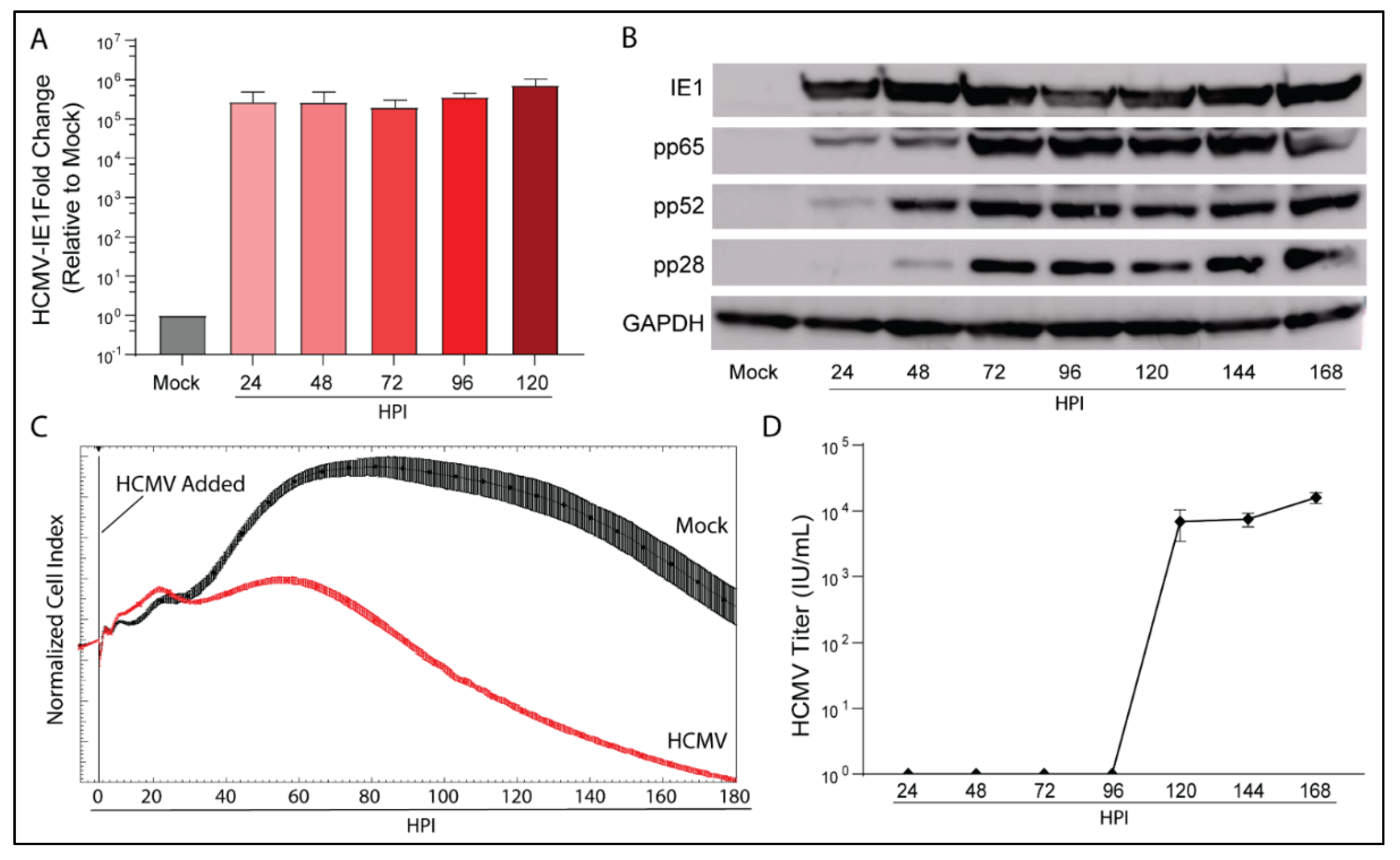
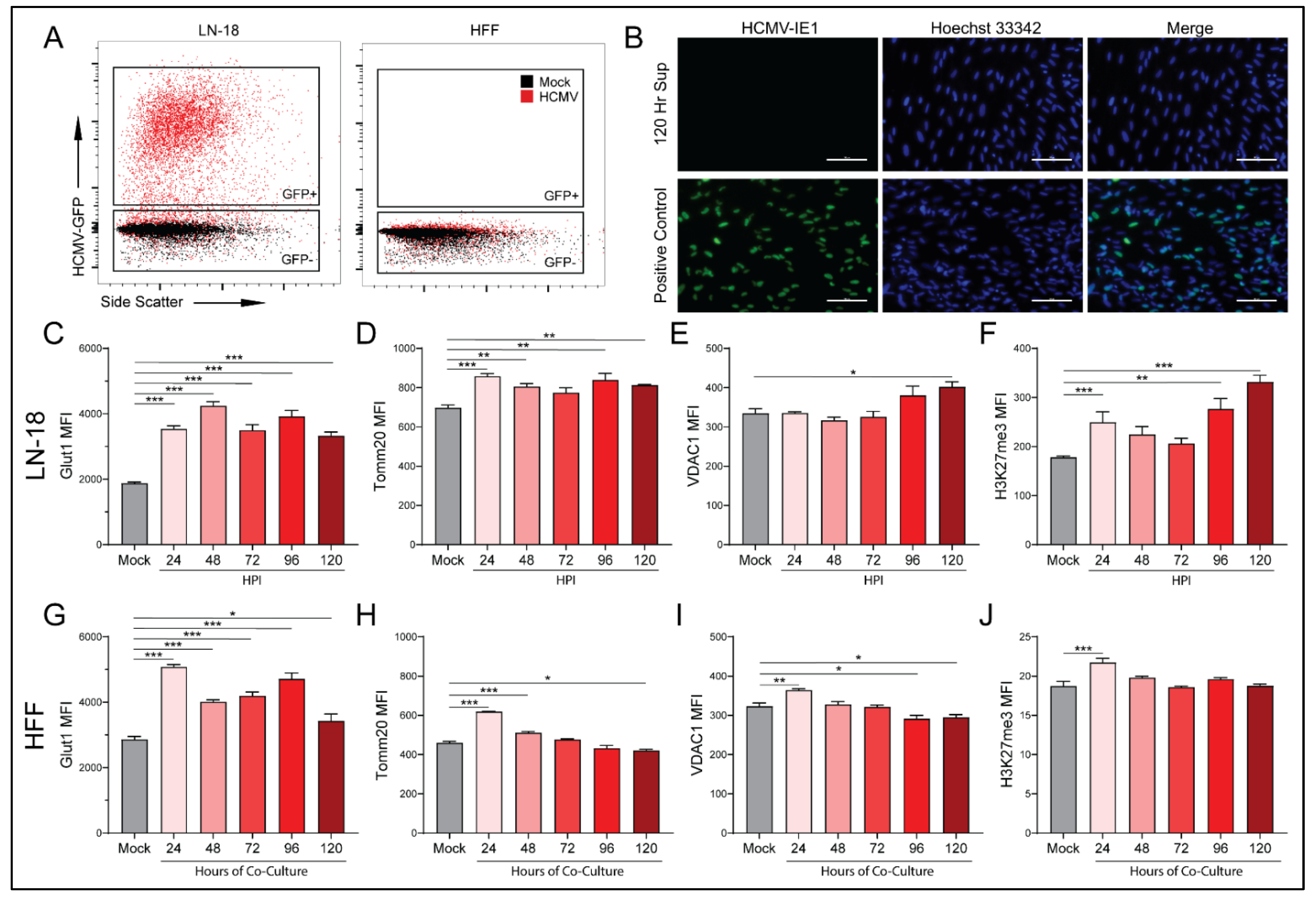
3.1. HCMV Infects and Replicates in a Human Glioblastoma Cell Line LN-18
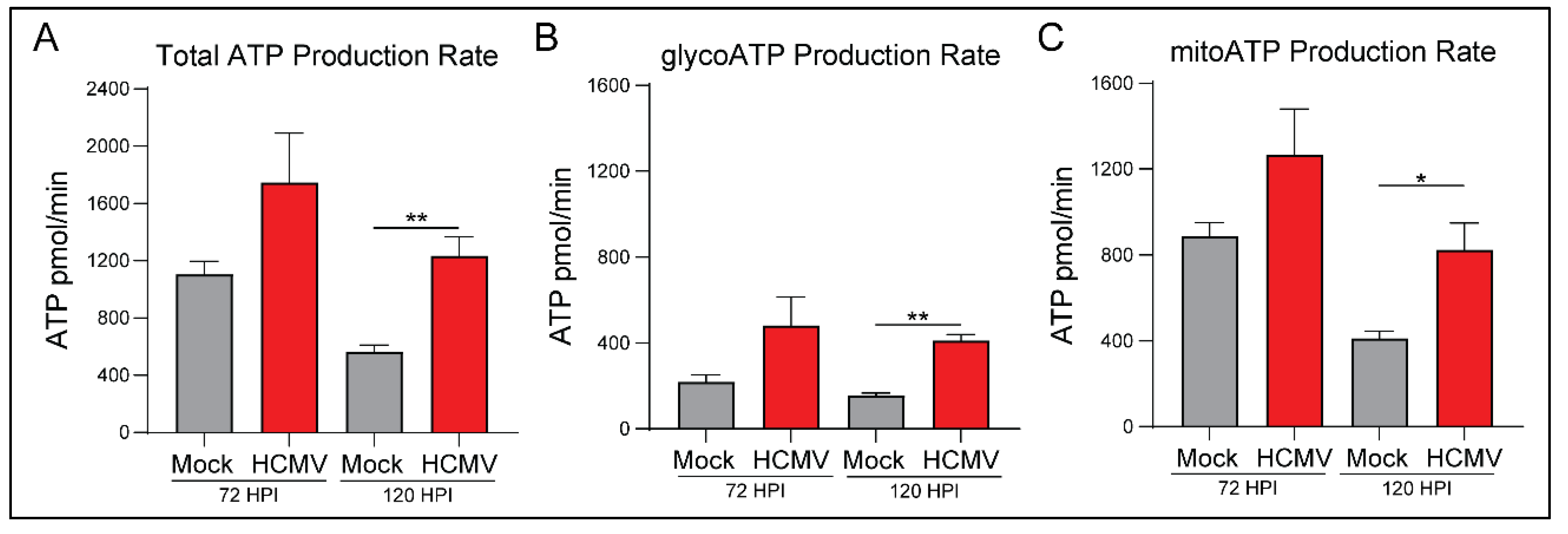
3.2. HCMV Infection Metabolically Reprograms LN-18 Cells
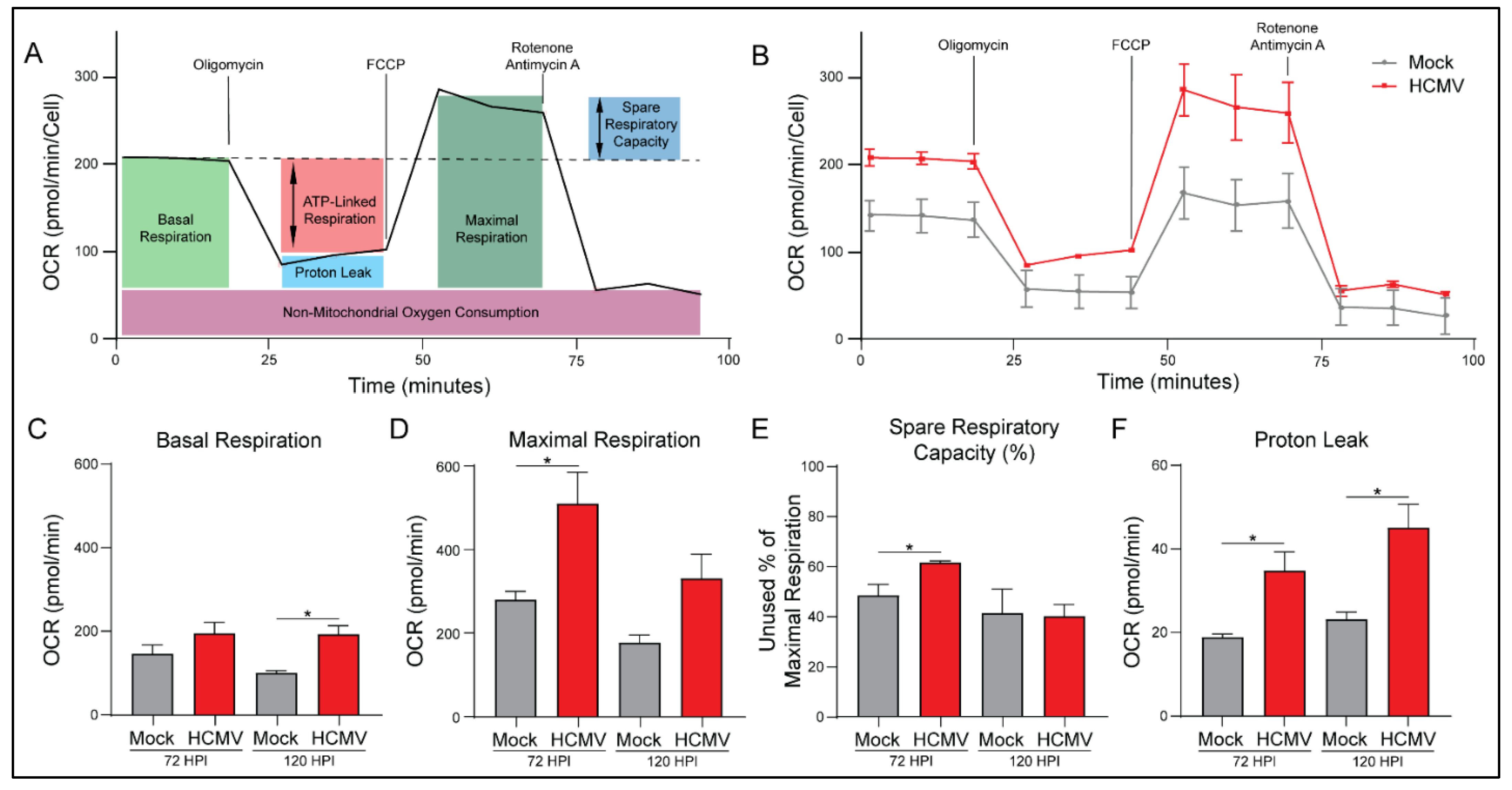
3.3. HCMV Infection Increases Oxidative Phosphorylation of Host Cells
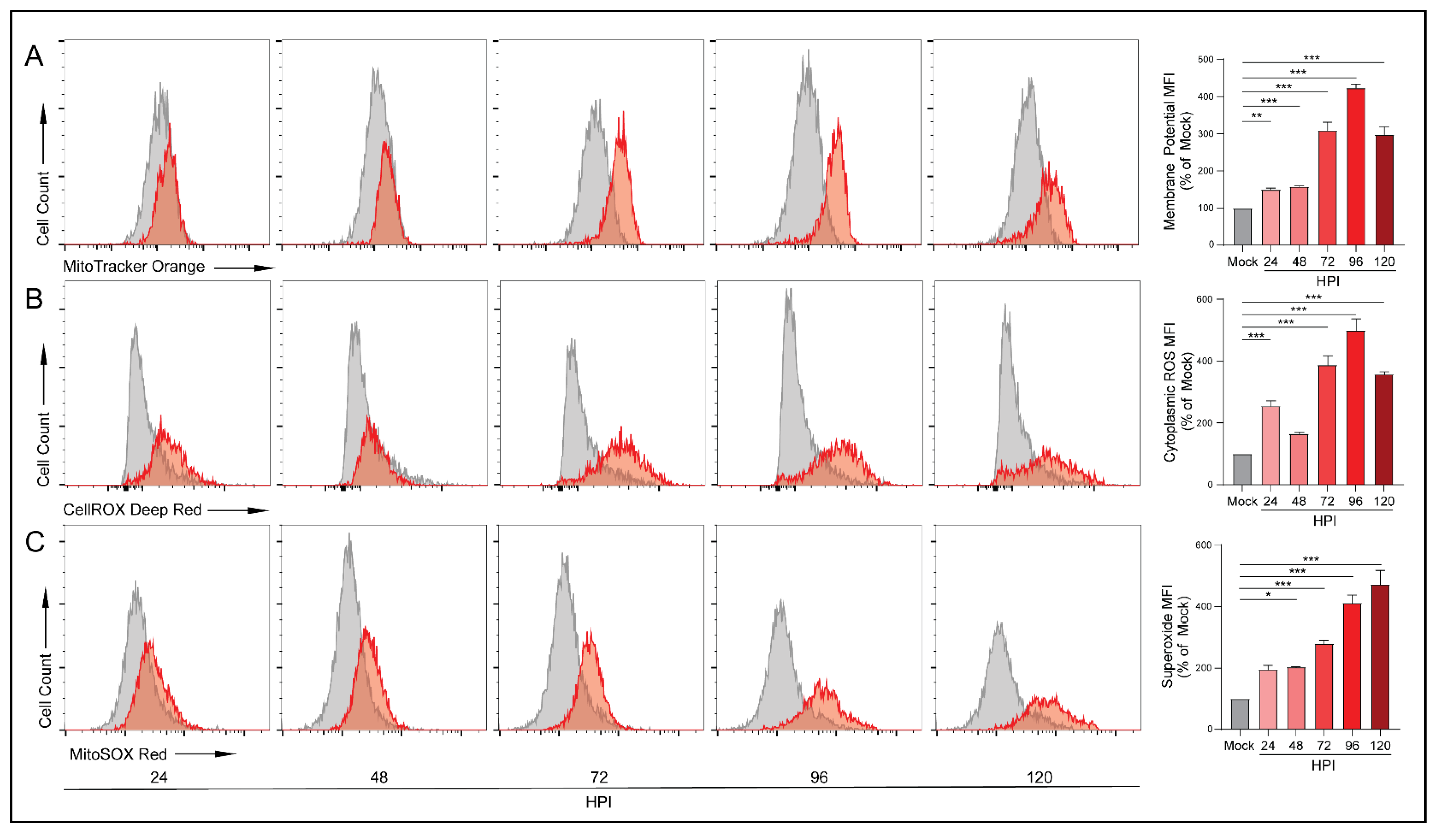
3.4. HCMV Infection Increases Mitochondrial Membrane Potential and Production of Reactive Oxygen Species
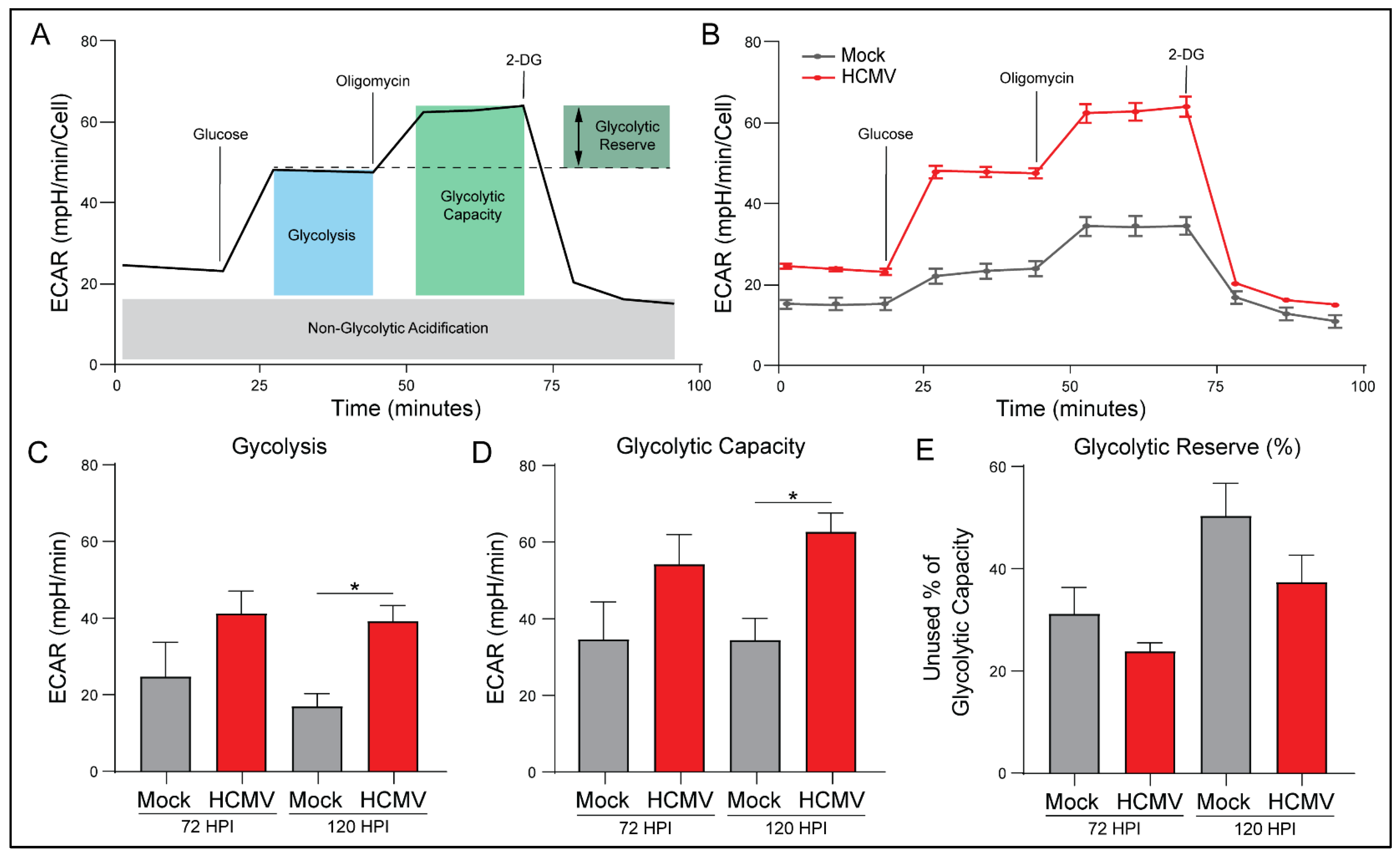
3.5. HCMV Infection Increases Glycolytic Rate and Capacity
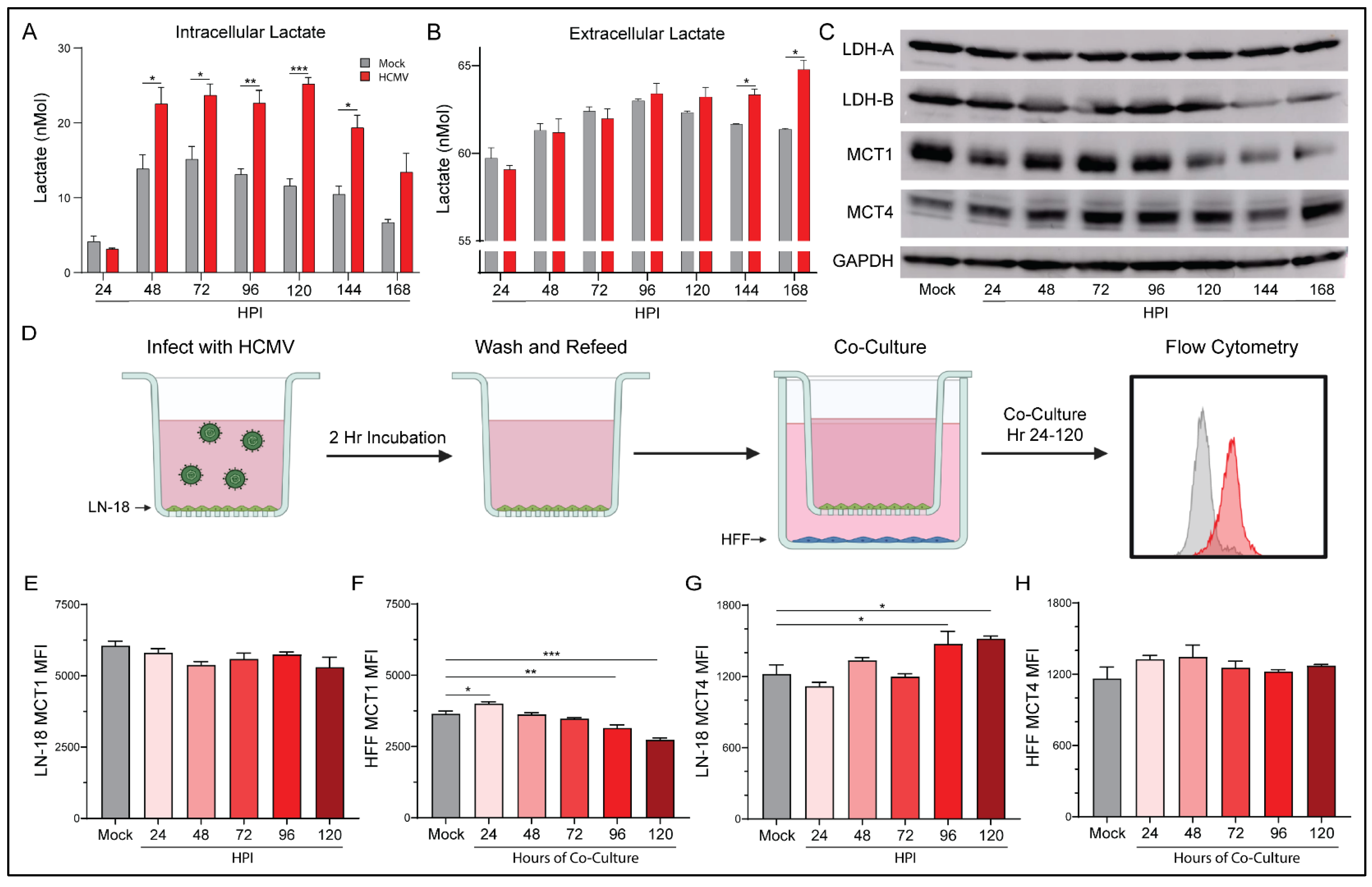
3.6. Elevated Aerobic Glycolysis following HCMV Infection Alters Lactate Production and Flux
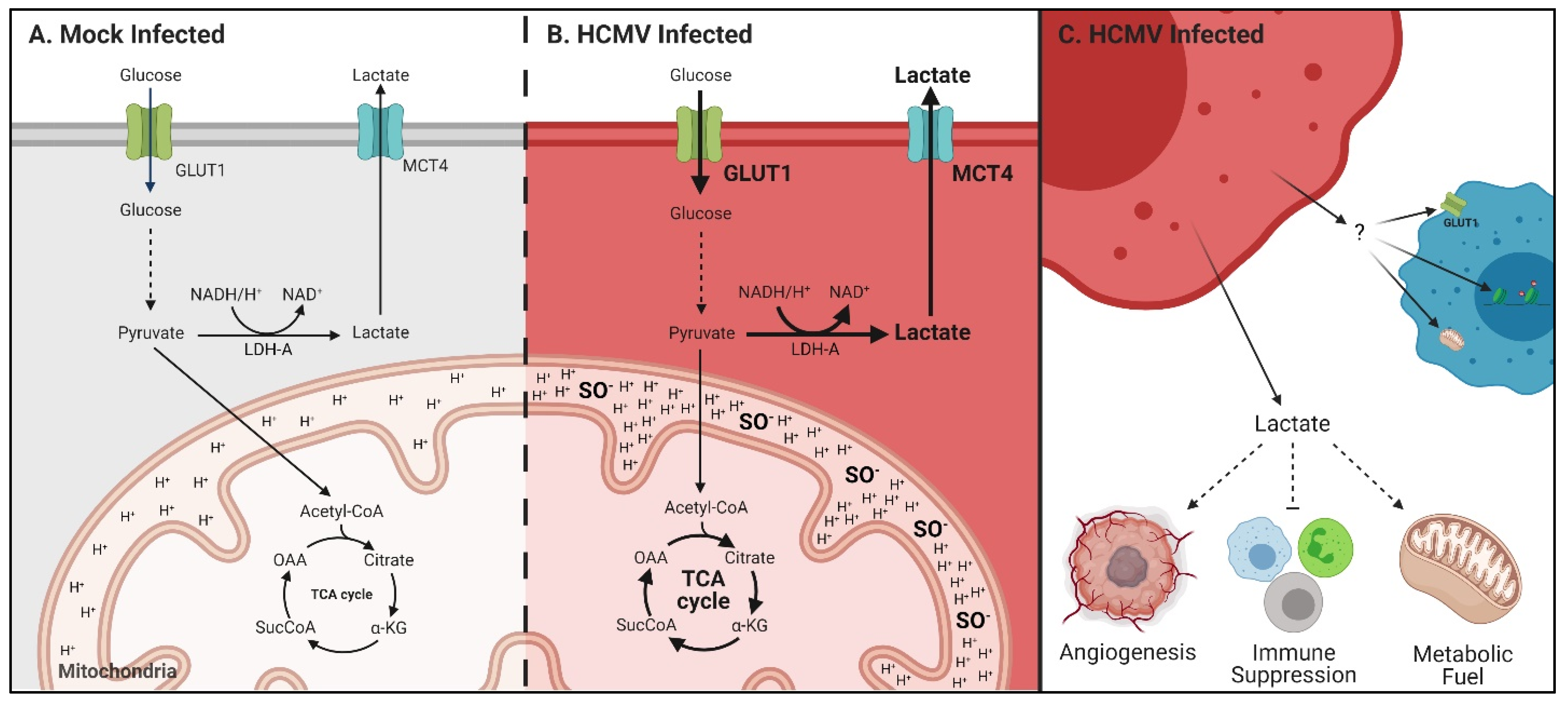
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Poon, M.T.C.; Sudlow, C.L.M.; Figueroa, J.D.; Brennan, P.M. Longer-term (>/= 2 years) survival in patients with glioblastoma in population-based studies pre- and post-2005: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 11622. [Google Scholar] [CrossRef]
- Fernandes, C.; Costa, A.; Osorio, L.; Lago, R.C.; Linhares, P.; Carvalho, B.; Caeiro, C. Current Standards of Care in Glioblastoma Therapy. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017; pp. 197–241. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Chang, Y.; Moore, P.S.; Weiss, R.A. Human oncogenic viruses: Nature and discovery. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [Green Version]
- Kofman, A.; Marcinkiewicz, L.; Dupart, E.; Lyshchev, A.; Martynov, B.; Ryndin, A.; Kotelevskaya, E.; Brown, J.; Schiff, D.; Abounader, R. The roles of viruses in brain tumor initiation and oncomodulation. J. NeuroOncol. 2011, 105, 451–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saddawi-Konefka, R.; Crawford, J.R. Chronic viral infection and primary central nervous system malignancy. J. Neuroimmune Pharmacol. 2010, 5, 387–403. [Google Scholar] [CrossRef] [Green Version]
- Cobbs, C.S.; Harkins, L.; Samanta, M.; Gillespie, G.Y.; Bharara, S.; King, P.H.; Nabors, L.B.; Cobbs, C.G.; Britt, W.J. Human cytomegalovirus infection and expression in human malignant glioma. Cancer Res. 2002, 62, 3347–3350. [Google Scholar] [PubMed]
- Maleki, F.; Sadigh, Z.A.; Sadeghi, F.; Muhammadnejad, A.; Farahmand, M.; Parvin, M.; Shirkoohi, R. Human cytomegalovirus infection in Iranian glioma patients correlates with aging and tumor aggressiveness. J. Med. Virol. 2020, 92, 1266–1276. [Google Scholar] [CrossRef]
- Mitchell, D.A.; Xie, W.; Schmittling, R.; Learn, C.; Friedman, A.; McLendon, R.E.; Sampson, J.H. Sensitive detection of human cytomegalovirus in tumors and peripheral blood of patients diagnosed with glioblastoma. Neuro Oncol. 2008, 10, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Scheurer, M.E.; Bondy, M.L.; Aldape, K.D.; Albrecht, T.; El-Zein, R. Detection of human cytomegalovirus in different histological types of gliomas. Acta NeuroPathol. 2008, 116, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Rahbar, A.; Orrego, A.; Peredo, I.; Dzabic, M.; Wolmer-Solberg, N.; Straat, K.; Stragliotto, G.; Soderberg-Naucler, C. Human cytomegalovirus infection levels in glioblastoma multiforme are of prognostic value for survival. J. Clin. Virol. 2013, 57, 36–42. [Google Scholar] [CrossRef]
- Libard, S.; Popova, S.N.; Amini, R.M.; Karja, V.; Pietilainen, T.; Hamalainen, K.M.; Sundstrom, C.; Hesselager, G.; Bergqvist, M.; Ekman, S.; et al. Human cytomegalovirus tegument protein pp65 is detected in all intra- and extra-axial brain tumours independent of the tumour type or grade. PLoS ONE 2014, 9, e108861. [Google Scholar] [CrossRef]
- Bhattacharjee, B.; Renzette, N.; Kowalik, T.F. Genetic analysis of cytomegalovirus in malignant gliomas. J. Virol. 2012, 86, 6815–6824. [Google Scholar] [CrossRef] [Green Version]
- Ranganathan, P.; Clark, P.A.; Kuo, J.S.; Salamat, M.S.; Kalejta, R.F. Significant association of multiple human cytomegalovirus genomic Loci with glioblastoma multiforme samples. J. Virol. 2012, 86, 854–864. [Google Scholar] [CrossRef] [Green Version]
- Lucas, K.G.; Bao, L.; Bruggeman, R.; Dunham, K.; Specht, C. The detection of CMV pp65 and IE1 in glioblastoma multiforme. J. NeuroOncol. 2011, 103, 231–238. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Chen, Y.Y.; Chen, W.G.; Diamond, D.J.; Mamelak, A.N.; Zaia, J.A.; Weiss, L.M. Lack of association of cytomegalovirus with human brain tumors. Mod. Pathol. 2005, 18, 838–843. [Google Scholar] [CrossRef] [PubMed]
- Baumgarten, P.; Michaelis, M.; Rothweiler, F.; Starzetz, T.; Rabenau, H.F.; Berger, A.; Jennewein, L.; Braczynski, A.K.; Franz, K.; Seifert, V.; et al. Human cytomegalovirus infection in tumor cells of the nervous system is not detectable with standardized pathologico-virological diagnostics. Neuro Oncol. 2014, 16, 1469–1477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stragliotto, G.; Pantalone, M.R.; Rahbar, A.; Bartek, J.; Soderberg-Naucler, C. Valganciclovir as Add-on to Standard Therapy in Glioblastoma Patients. Clin. Cancer Res. 2020, 26, 4031–4039. [Google Scholar] [CrossRef] [PubMed]
- Stragliotto, G.; Rahbar, A.; Solberg, N.W.; Lilja, A.; Taher, C.; Orrego, A.; Bjurman, B.; Tammik, C.; Skarman, P.; Peredo, I.; et al. Effects of valganciclovir as an add-on therapy in patients with cytomegalovirus-positive glioblastoma: A randomized, double-blind, hypothesis-generating study. Int. J. Cancer 2013, 133, 1204–1213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staras, S.A.; Dollard, S.C.; Radford, K.W.; Flanders, W.D.; Pass, R.F.; Cannon, M.J. Seroprevalence of cytomegalovirus infection in the United States, 1988–1994. Clin. Infect. Dis. 2006, 43, 1143–1151. [Google Scholar] [CrossRef] [PubMed]
- Mustakangas, P.; Sarna, S.; Ammala, P.; Muttilainen, M.; Koskela, P.; Koskiniemi, M. Human cytomegalovirus seroprevalence in three socioeconomically different urban areas during the first trimester: A population-based cohort study. Int. J. Epidemiol. 2000, 29, 587–591. [Google Scholar] [CrossRef]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Saha, I.; Narayanan, A.; Narayanan, S.; Takaoka, A.; Kumar, N.S.; Tailor, P.; Kumar, H. Essential role of HCMV deubiquitinase in promoting oncogenesis by targeting anti-viral innate immune signaling pathways. Cell Death Dis. 2017, 8, e3078. [Google Scholar] [CrossRef] [Green Version]
- Siew, V.K.; Duh, C.Y.; Wang, S.K. Human cytomegalovirus UL76 induces chromosome aberrations. J. Biomed. Sci. 2009, 16, 107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, C.S.; Ljunggren, H.G.; Achour, A. Immune modulation by the human cytomegalovirus-encoded molecule UL18, a mystery yet to be solved. J. Immunol. 2008, 180, 19–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munger, J.; Bennett, B.D.; Parikh, A.; Feng, X.J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog 2006, 2, e132. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J. Virol. 2011, 85, 1573–1580. [Google Scholar] [CrossRef] [Green Version]
- Kaarbo, M.; Ager-Wick, E.; Osenbroch, P.O.; Kilander, A.; Skinnes, R.; Muller, F.; Eide, L. Human cytomegalovirus infection increases mitochondrial biogenesis. Mitochondrion 2011, 11, 935–945. [Google Scholar] [CrossRef]
- Combs, J.A.; Norton, E.B.; Saifudeen, Z.R.; Bentrup, K.H.Z.; Katakam, P.V.; Morris, C.A.; Myers, L.; Kaur, A.; Sullivan, D.E.; Zwezdaryk, K.J. Human Cytomegalovirus Alters Host Cell Mitochondrial Function during Acute Infection. J. Virol. 2020, 94, e01183-19. [Google Scholar] [CrossRef] [Green Version]
- Betsinger, C.N.; Jankowski, C.S.R.; Hofstadter, W.A.; Federspiel, J.D.; Otter, C.J.; Jean Beltran, P.M.; Cristea, I.M. The human cytomegalovirus protein pUL13 targets mitochondrial cristae architecture to increase cellular respiration during infection. Proc. Natl. Acad. Sci. USA 2021, 118, e2101675118. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Warburg, O. On respiratory impairment in cancer cells. Science 1956, 124, 269–270. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britt, W.J. Human cytomegalovirus: Propagation, quantification, and storage. Curr. Protoc. MicroBiol. 2010, 18, 14E.3.1–14E.3.17. [Google Scholar] [CrossRef] [PubMed]
- Krenzlin, H.; Behera, P.; Lorenz, V.; Passaro, C.; Zdioruk, M.; Nowicki, M.O.; Grauwet, K.; Zhang, H.; Skubal, M.; Ito, H.; et al. Cytomegalovirus promotes murine glioblastoma growth via pericyte recruitment and angiogenesis. J. Clin. Investig. 2019, 129, 1671–1683. [Google Scholar] [CrossRef] [Green Version]
- Martin, K.R.; Barrett, J.C. Reactive oxygen species as double-edged swords in cellular processes: Low-dose cell signaling versus high-dose toxicity. Hum. Exp. Toxicol. 2002, 21, 71–75. [Google Scholar] [CrossRef]
- Rodriguez-Sanchez, I.; Schafer, X.L.; Monaghan, M.; Munger, J. The Human Cytomegalovirus UL38 protein drives mTOR-independent metabolic flux reprogramming by inhibiting TSC2. PLoS Pathog. 2019, 15, e1007569. [Google Scholar] [CrossRef]
- Ezponda, T.; Licht, J.D. Molecular pathways: Deregulation of histone h3 lysine 27 methylation in cancer-different paths, same destination. Clin. Cancer Res. 2014, 20, 5001–5008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngollo, M.; Lebert, A.; Daures, M.; Judes, G.; Rifai, K.; Dubois, L.; Kemeny, J.L.; Penault-Llorca, F.; Bignon, Y.J.; Guy, L.; et al. Global analysis of H3K27me3 as an epigenetic marker in prostate cancer progression. BMC Cancer 2017, 17, 261. [Google Scholar] [CrossRef]
- Fang, S.; Shen, Y.; Chen, B.; Wu, Y.; Jia, L.; Li, Y.; Zhu, Y.; Yan, Y.; Li, M.; Chen, R.; et al. H3K27me3 induces multidrug resistance in small cell lung cancer by affecting HOXA1 DNA methylation via regulation of the lncRNA HOTAIR. Ann. Transl. Med. 2018, 6, 440. [Google Scholar] [CrossRef]
- Lin, B.; Lee, H.; Yoon, J.G.; Madan, A.; Wayner, E.; Tonning, S.; Hothi, P.; Schroeder, B.; Ulasov, I.; Foltz, G.; et al. Global analysis of H3K4me3 and H3K27me3 profiles in glioblastoma stem cells and identification of SLC17A7 as a bivalent tumor suppressor gene. Oncotarget 2015, 6, 5369–5381. [Google Scholar] [CrossRef] [Green Version]
- Lawler, S.E. Cytomegalovirus and glioblastoma; controversies and opportunities. J. NeuroOncol. 2015, 123, 465–471. [Google Scholar] [CrossRef]
- Stragliotto, G.; Pantalone, M.R.; Rahbar, A.; Soderberg-Naucler, C. Valganciclovir as Add-On to Standard Therapy in Secondary Glioblastoma. Microorganisms 2020, 8, 1471. [Google Scholar] [CrossRef] [PubMed]
- Marin-Valencia, I.; Yang, C.; Mashimo, T.; Cho, S.; Baek, H.; Yang, X.L.; Rajagopalan, K.N.; Maddie, M.; Vemireddy, V.; Zhao, Z.; et al. Analysis of tumor metabolism reveals mitochondrial glucose oxidation in genetically diverse human glioblastomas in the mouse brain in vivo. Cell Metab. 2012, 15, 827–837. [Google Scholar] [CrossRef] [Green Version]
- Maher, E.A.; Marin-Valencia, I.; Bachoo, R.M.; Mashimo, T.; Raisanen, J.; Hatanpaa, K.J.; Jindal, A.; Jeffrey, F.M.; Choi, C.; Madden, C.; et al. Metabolism of [U-13 C]glucose in human brain tumors in vivo. NMR Biomed. 2012, 25, 1234–1244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sighel, D.; Notarangelo, M.; Aibara, S.; Re, A.; Ricci, G.; Guida, M.; Soldano, A.; Adami, V.; Ambrosini, C.; Broso, F.; et al. Inhibition of mitochondrial translation suppresses glioblastoma stem cell growth. Cell Rep. 2021, 35, 109024. [Google Scholar] [CrossRef]
- Kumari, S.; Badana, A.K.; Murali Mohan, G.; Shailender, G.; Malla, R. Reactive Oxygen Species: A Key Constituent in Cancer Survival. Biomark Insights 2018, 13, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Liou, G.Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.R.; Yang, K.S.; Kwon, J.; Lee, C.; Jeong, W.; Rhee, S.G. Reversible inactivation of the tumor suppressor PTEN by H2O2. J. Biol. Chem. 2002, 277, 20336–20342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combs, J.A.; Monk, C.H.; Harrison, M.A.A.; Norton, E.B.; Morris, C.A.; Sullivan, D.E.; Zwezdaryk, K.J. Inhibiting cytomegalovirus replication through targeting the host electron transport chain. Antiviral Res. 2021, 194, 105159. [Google Scholar] [CrossRef]
- Sanzey, M.; Abdul Rahim, S.A.; Oudin, A.; Dirkse, A.; Kaoma, T.; Vallar, L.; Herold-Mende, C.; Bjerkvig, R.; Golebiewska, A.; Niclou, S.P. Comprehensive analysis of glycolytic enzymes as therapeutic targets in the treatment of glioblastoma. PLoS ONE 2015, 10, e0123544. [Google Scholar] [CrossRef]
- de Wit, R.H.; Mujic-Delic, A.; van Senten, J.R.; Fraile-Ramos, A.; Siderius, M.; Smit, M.J. Human cytomegalovirus encoded chemokine receptor US28 activates the HIF-1alpha/PKM2 axis in glioblastoma cells. Oncotarget 2016, 7, 67966–67985. [Google Scholar] [CrossRef] [Green Version]
- Hjelmeland, A.B.; Wu, Q.; Heddleston, J.M.; Choudhary, G.S.; MacSwords, J.; Lathia, J.D.; McLendon, R.; Lindner, D.; Sloan, A.; Rich, J.N. Acidic stress promotes a glioma stem cell phenotype. Cell Death Differ. 2011, 18, 829–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, S.W.; Lin, H.J.; Liu, Y.S.; Yang, L.Y.; Lu, D.Y. Monocarboxylate Transporter 4 Regulates Glioblastoma Motility and Monocyte Binding Ability. Cancers 2020, 12, 380. [Google Scholar] [CrossRef] [Green Version]
- Cuyas, E.; Corominas-Faja, B.; Menendez, J.A. The nutritional phenome of EMT-induced cancer stem-like cells. Oncotarget 2014, 5, 3970–3982. [Google Scholar] [CrossRef] [Green Version]
- de la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef] [Green Version]
- Nakano, I.; Garnier, D.; Minata, M.; Rak, J. Extracellular vesicles in the biology of brain tumour stem cells—Implications for inter-cellular communication, therapy and biomarker development. Semin. Cell Dev. Biol. 2015, 40, 17–26. [Google Scholar] [CrossRef]
- Seo, J.Y.; Cresswell, P. Viperin regulates cellular lipid metabolism during human cytomegalovirus infection. PLoS Pathog 2013, 9, e1003497. [Google Scholar] [CrossRef]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro Oncol. 2017, 19, 43–54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.O.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [Green Version]
- Ullah, M.S.; Davies, A.J.; Halestrap, A.P. The plasma membrane lactate transporter MCT4, but not MCT1, is up-regulated by hypoxia through a HIF-1alpha-dependent mechanism. J. Biol. Chem. 2006, 281, 9030–9037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Clark, P.A.; Kuo, J.S.; Kalejta, R.F. Human Cytomegalovirus-Infected Glioblastoma Cells Display Stem Cell-Like Phenotypes. mSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harrison, M.A.A.; Hochreiner, E.M.; Benjamin, B.P.; Lawler, S.E.; Zwezdaryk, K.J. Metabolic Reprogramming of Glioblastoma Cells during HCMV Infection Induces Secretome-Mediated Paracrine Effects in the Microenvironment. Viruses 2022, 14, 103. https://doi.org/10.3390/v14010103
Harrison MAA, Hochreiner EM, Benjamin BP, Lawler SE, Zwezdaryk KJ. Metabolic Reprogramming of Glioblastoma Cells during HCMV Infection Induces Secretome-Mediated Paracrine Effects in the Microenvironment. Viruses. 2022; 14(1):103. https://doi.org/10.3390/v14010103
Chicago/Turabian StyleHarrison, Mark A. A., Emily M. Hochreiner, Brooke P. Benjamin, Sean E. Lawler, and Kevin J. Zwezdaryk. 2022. "Metabolic Reprogramming of Glioblastoma Cells during HCMV Infection Induces Secretome-Mediated Paracrine Effects in the Microenvironment" Viruses 14, no. 1: 103. https://doi.org/10.3390/v14010103
APA StyleHarrison, M. A. A., Hochreiner, E. M., Benjamin, B. P., Lawler, S. E., & Zwezdaryk, K. J. (2022). Metabolic Reprogramming of Glioblastoma Cells during HCMV Infection Induces Secretome-Mediated Paracrine Effects in the Microenvironment. Viruses, 14(1), 103. https://doi.org/10.3390/v14010103