
Replication and Spread of Oncolytic Herpes Simplex Virus in Solid Tumors


Abstract
:1. Introduction
2. Infection and Spread of HSV-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Factor Types | Factors | Function | References |
|---|---|---|---|
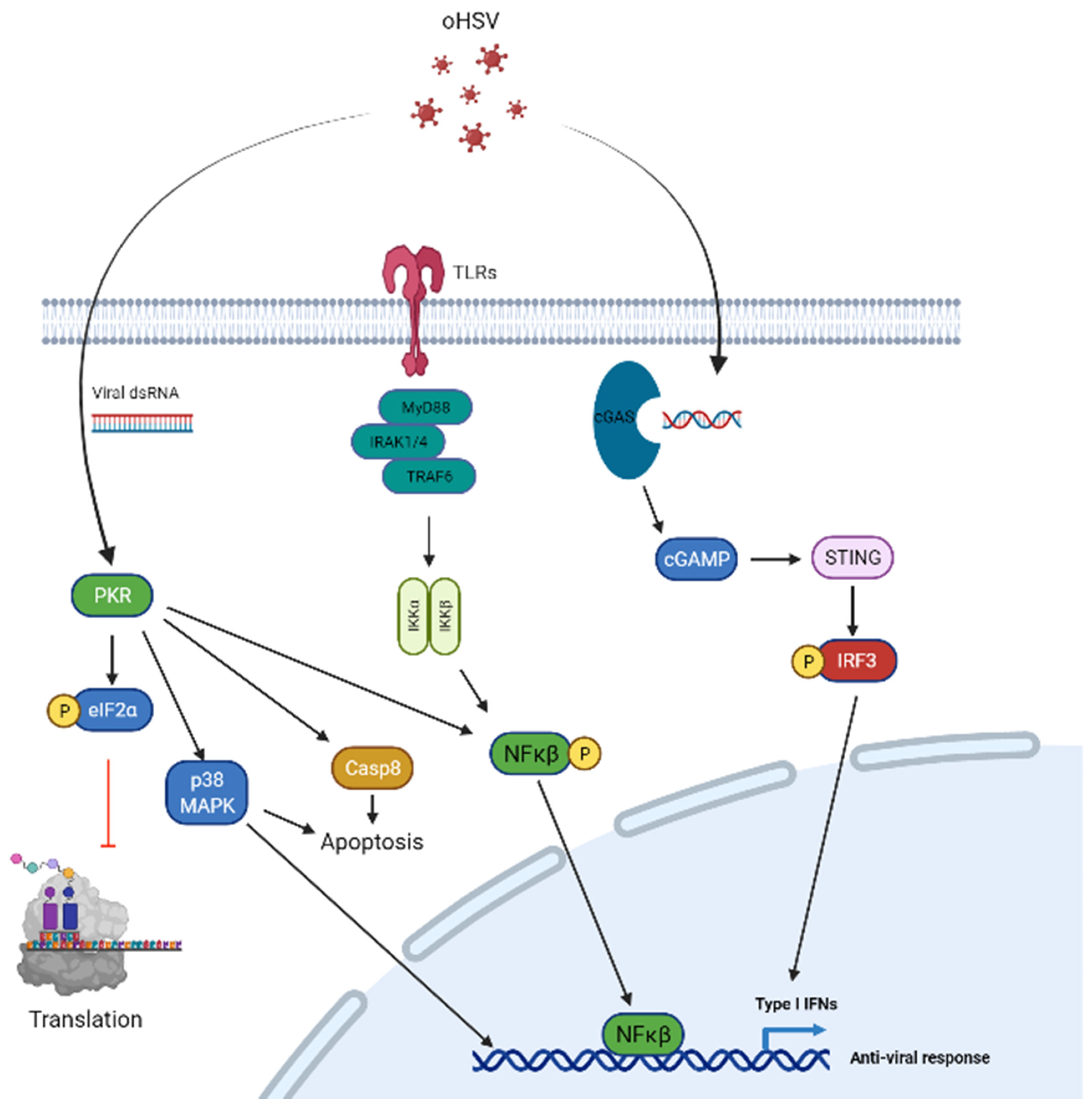
| Cancer cell intrinsic factors | PKR | Block cellular protein translation | [21,22,23,24,25,26,27,28,29,30,31] |
| cGAS-STING | Induce ISGs to limit virus infection | [32,33,34,35] | |
| Type I interferon | Activation of JAK–STAT1 and transcription of ISGs to inhibit virus infection | [36,37] | |
| TLR | Sense viral infection and activate NF-κB | [38,39] | |
| Virus genes | UL23 | Encodes thymidine kinase; Viral enzymes involved in nucleotide metabolism | [40] |
| RL1 | Virulence for neuron infection and latency | [41,42,43,44,45,46,47,48,49,50,51,52,53] | |
| ICP4 | Immediate early (IE) protein that is required for early and late gene transcription | [54,55,56,57,58,59] | |
| ICP6 | Encodes ribonucleotide reductase; converts ribonucleotides to deoxy-ribonucleotides; essential for HSV viral DNA replication | [60,61,62,63] | |
| ICP47 | Viral protein downregulating antigen presentation activity by inhibiting transporter associated with antigen processing protein (TAP) function | [63] |
3. Cancer Cell Intrinsic Barriers That Limit oHSV-1
4. HSV-1 Encoded Genes That Are Manipulated to Generate Tumor Specific Oncolytic HSV
4.1. UL23
4.2. RL1
4.3. ICP4
4.4. ICP6
4.5. ICP47
4.6. Entry Tropism Modification
5. Modulation of Cellular Signaling to Synergize with oHSV Therapy
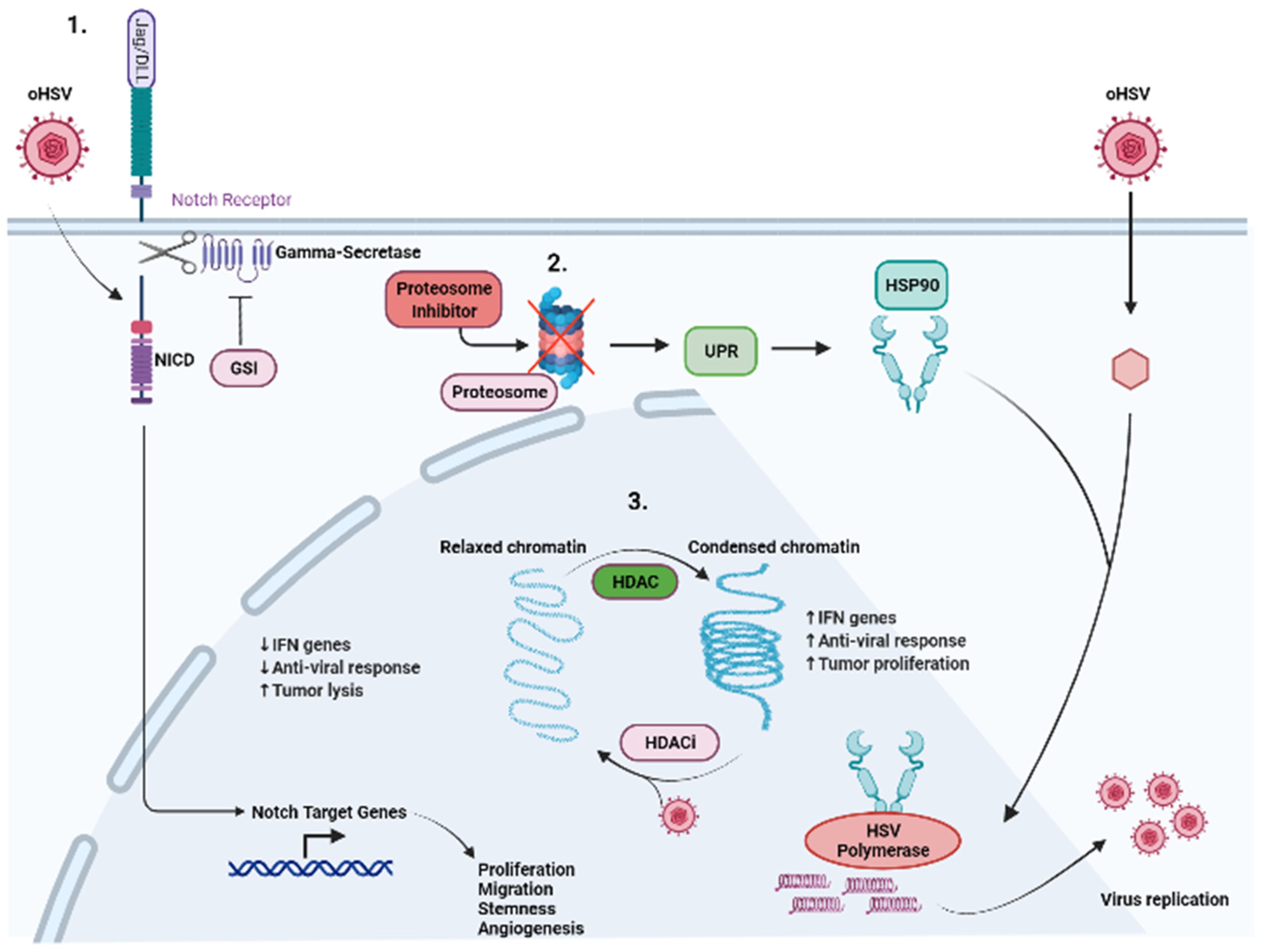
5.1. Proteasome Inhibitor
5.2. NOTCH
5.3. HDAC
5.4. cGAS-STING
5.5. DNA Damage Response (DDR) Inducer
5.6. MEK Inhibitor
5.7. PTEN/PI3K/AKT Signaling
6. Modulation of Tumor ECM to Enhance oHSV Therapy
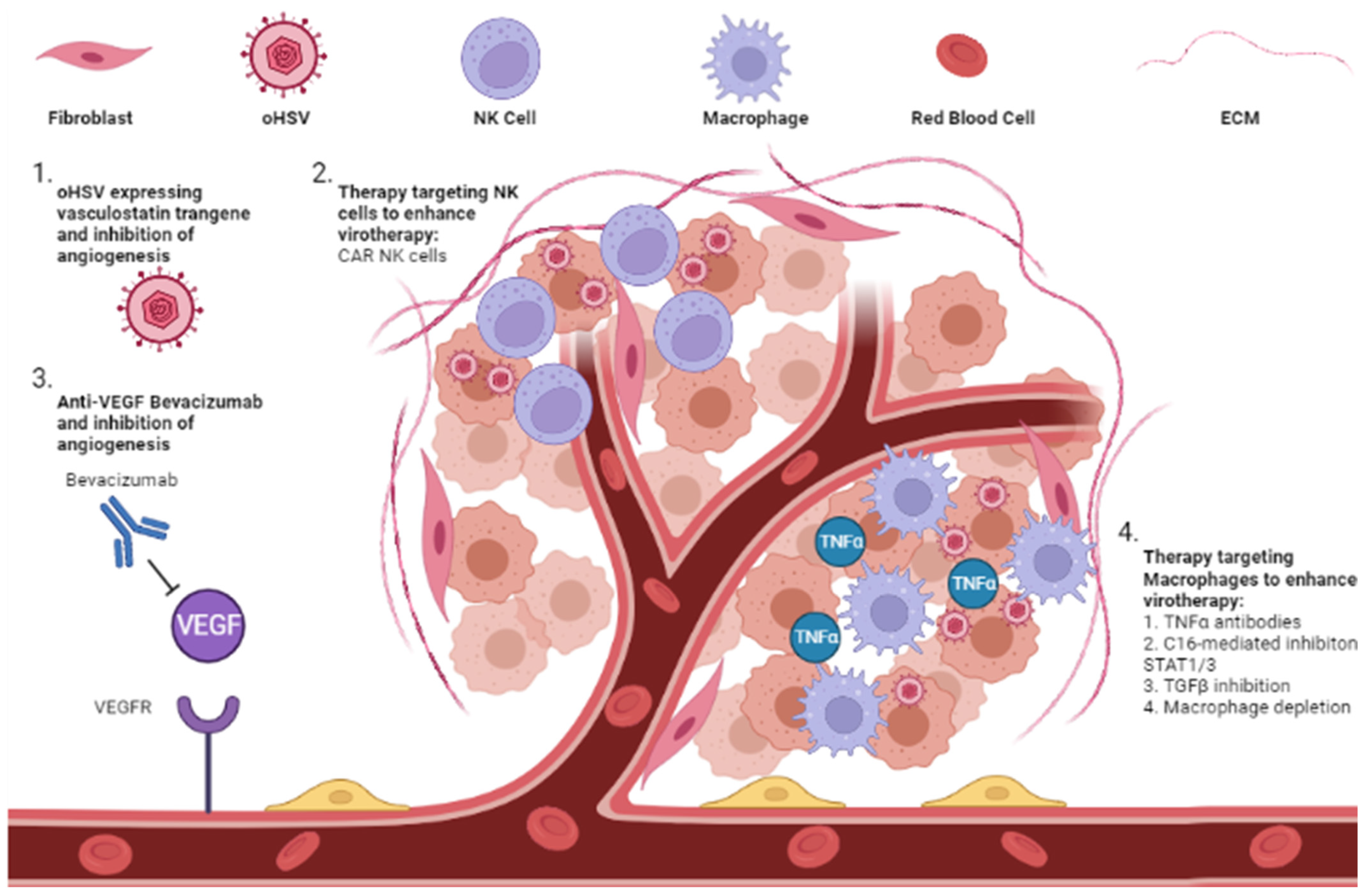
6.1. Anti-Angiogenic Inhibitor
6.2. Extracellular Immune Cells
7. Strategies to Improve oHSV Delivery and Spread
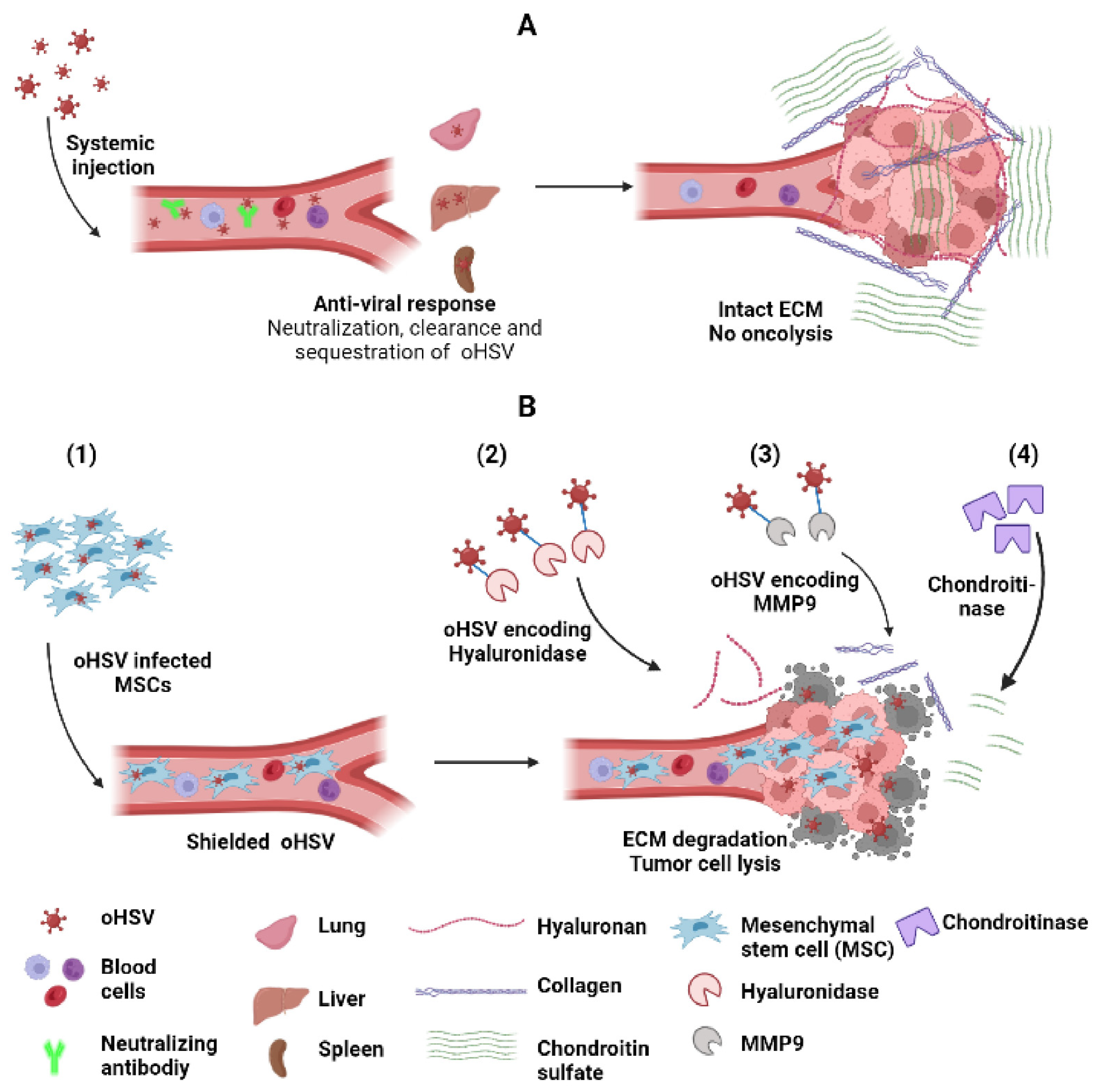
7.1. Carrier Cells
7.2. Increasing oHSV Spread into the Tumor Cells
8. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Goodpasture, E.W.; Teague, O. Transmission of the Virus of Herpes Febrilis along Nerves in experimentally infected Rabbits. J. Med. Res. 1923, 44, 139–184.7. [Google Scholar] [PubMed]
- Bradley, H.; Markowitz, L.E.; Gibson, T.; McQuillan, G.M. Seroprevalence of herpes simplex virus types 1 and 2—United States, 1999–2010. J. Infect. Dis. 2014, 209, 325–333. [Google Scholar] [CrossRef] [PubMed]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global and Regional Estimates of Prevalent and Incident Herpes Simplex Virus Type 1 Infections in 2012. PLoS ONE 2015, 10, e0140765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodpasture, E.W.; Teague, O. Experimental Production of Herpetic Lesions in Organs and Tissues of the Rabbit. J. Med. Res. 1923, 44, 121–138.5. [Google Scholar]
- Andtbacka, R.H.; Kaufman, H.L.; Collichio, F.; Amatruda, T.; Senzer, N.; Chesney, J.; Delman, K.A.; Spitler, L.E.; Puzanov, I.; Agarwala, S.S.; et al. Talimogene Laherparepvec Improves Durable Response Rate in Patients With Advanced Melanoma. J. Clin. Oncol. 2015, 33, 2780–2788. [Google Scholar] [CrossRef]
- Moesta, A.K.; Cooke, K.; Piasecki, J.; Mitchell, P.; Rottman, J.B.; Fitzgerald, K.; Zhan, J.; Yang, B.; Le, T.; Belmontes, B.; et al. Local Delivery of OncoVEX(mGM-CSF) Generates Systemic Antitumor Immune Responses Enhanced by Cytotoxic T-Lymphocyte-Associated Protein Blockade. Clin. Cancer Res. 2017, 23, 6190–6202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrington, K.J.; Kong, A.; Mach, N.; Chesney, J.A.; Fernandez, B.C.; Rischin, D.; Cohen, E.E.W.; Radcliffe, H.S.; Gumuscu, B.; Cheng, J.; et al. Talimogene Laherparepvec and Pembrolizumab in Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (MASTERKEY-232): A Multicenter, Phase 1b Study. Clin. Cancer Res. 2020, 26, 5153–5161. [Google Scholar] [CrossRef] [PubMed]
- Chesney, J.; Puzanov, I.; Collichio, F.; Singh, P.; Milhem, M.M.; Glaspy, J.; Hamid, O.; Ross, M.; Friedlander, P.; Garbe, C.; et al. Randomized, Open-Label Phase II Study Evaluating the Efficacy and Safety of Talimogene Laherparepvec in Combination With Ipilimumab Versus Ipilimumab Alone in Patients With Advanced, Unresectable Melanoma. J. Clin. Oncol. 2018, 36, 1658–1667. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.; Hutzen, B.; Wedekind, M.F.; Cripe, T.P. Oncolytic virus and PD-1/PD-L1 blockade combination therapy. Oncolytic Virother 2018, 7, 65–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mettenleiter, T.C.; Zsak, L.; Zuckermann, F.; Sugg, N.; Kern, H.; Ben-Porat, T. Interaction of glycoprotein gIII with a cellular heparinlike substance mediates adsorption of pseudorabies virus. J. Virol. 1990, 64, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- WuDunn, D.; Spear, P.G. Initial interaction of herpes simplex virus with cells is binding to heparan sulfate. J. Virol. 1989, 63, 52–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, B.C.; Visalli, R.J.; Susmarski, N.; Brandt, C.R.; Spear, P.G. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulphate and glycoprotein B. J. Gen. Virol 1994, 75, 1211–1222. [Google Scholar] [CrossRef] [PubMed]
- Geraghty, R.J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar] [CrossRef]
- Shukla, D.; Liu, J.; Blaiklock, P.; Shworak, N.W.; Bai, X.; Esko, J.D.; Cohen, G.H.; Eisenberg, R.J.; Rosenberg, R.D.; Spear, P.G. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999, 99, 13–22. [Google Scholar] [CrossRef] [Green Version]
- Chowdary, T.K.; Cairns, T.M.; Atanasiu, D.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 2010, 17, 882–888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luxton, G.W.; Haverlock, S.; Coller, K.E.; Antinone, S.E.; Pincetic, A.; Smith, G.A. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 5832–5837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jovasevic, V.; Liang, L.; Roizman, B. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 2008, 82, 3311–3319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Granzow, H.; Weiland, F.; Jons, A.; Klupp, B.G.; Karger, A.; Mettenleiter, T.C. Ultrastructural analysis of the replication cycle of pseudorabies virus in cell culture: A reassessment. J. Virol. 1997, 71, 2072–2082. [Google Scholar] [CrossRef] [Green Version]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef]
- Ito, T.; Yang, M.; May, W.S. RAX, a cellular activator for double-stranded RNA-dependent protein kinase during stress signaling. J. Biol. Chem. 1999, 274, 15427–15432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, R.C.; Sen, G.C. PACT, a protein activator of the interferon-induced protein kinase, PKR. EMBO J. 1998, 17, 4379–4390. [Google Scholar] [CrossRef]
- Galabru, J.; Hovanessian, A. Autophosphorylation of the protein kinase dependent on double-stranded RNA. J. Biol. Chem. 1987, 262, 15538–15544. [Google Scholar] [CrossRef]
- Rhoads, R.E. Regulation of eukaryotic protein synthesis by initiation factors. J. Biol. Chem. 1993, 268, 3017–3020. [Google Scholar] [CrossRef]
- Balachandran, S.; Roberts, P.C.; Brown, L.E.; Truong, H.; Pattnaik, A.K.; Archer, D.R.; Barber, G.N. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity 2000, 13, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Stojdl, D.F.; Abraham, N.; Knowles, S.; Marius, R.; Brasey, A.; Lichty, B.D.; Brown, E.G.; Sonenberg, N.; Bell, J.C. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J. Virol. 2000, 74, 9580–9585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Gross, M.; Roizman, B. The gamma (1) 34.5 protein of herpes simplex virus 1 complexes with protein phosphatase 1alpha to dephosphorylate the alpha subunit of the eukaryotic translation initiation factor 2 and preclude the shutoff of protein synthesis by double-stranded RNA-activated protein kinase. Proc. Natl. Acad. Sci. USA 1997, 94, 843–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, B.; Gross, M.; Roizman, B. The gamma134.5 protein of herpes simplex virus 1 has the structural and functional attributes of a protein phosphatase 1 regulatory subunit and is present in a high molecular weight complex with the enzyme in infected cells. J. Biol. Chem. 1998, 273, 20737–20743. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cassady, K.A.; Gross, M. The herpes simplex virus type 1 U(S)11 protein interacts with protein kinase R in infected cells and requires a 30-amino-acid sequence adjacent to a kinase substrate domain. J. Virol. 2002, 76, 2029–2035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, G.A.; Khoo, D.; Mohr, I.; Sen, G.C. Inhibition of PACT-mediated activation of PKR by the herpes simplex virus type 1 Us11 protein. J. Virol. 2002, 76, 11054–11064. [Google Scholar] [CrossRef] [Green Version]
- Dauber, B.; Pelletier, J.; Smiley, J.R. The herpes simplex virus 1 vhs protein enhances translation of viral true late mRNAs and virus production in a cell type-dependent manner. J. Virol. 2011, 85, 5363–5373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, H.; Sun, H.; You, F.; Sun, W.; Zhou, X.; Chen, L.; Yang, J.; Wang, Y.; Tang, H.; Guan, Y.; et al. Activation of STAT6 by STING is critical for antiviral innate immunity. Cell 2011, 147, 436–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gui, X.; Yang, H.; Li, T.; Tan, X.; Shi, P.; Li, M.; Du, F.; Chen, Z.J. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019, 567, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef] [PubMed]
- Teijaro, J.R.; Ng, C.; Lee, A.M.; Sullivan, B.M.; Sheehan, K.C.; Welch, M.; Schreiber, R.D.; de la Torre, J.C.; Oldstone, M.B. Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013, 340, 207–211. [Google Scholar] [CrossRef] [Green Version]
- Perez de Diego, R.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; Plancoulaine, S.; Picard, C.; Herman, M.; Cardon, A.; Durandy, A.; Bustamante, J.; et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity 2010, 33, 400–411. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Liu, S.; Chen, Z.J. SnapShot: Pathways of antiviral innate immunity. Cell 2010, 140, 436–436.e2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martuza, R.L.; Malick, A.; Markert, J.M.; Ruffner, K.L.; Coen, D.M. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science 1991, 252, 854–856. [Google Scholar] [CrossRef] [PubMed]
- Kesari, S.; Lee, V.M.; Brown, S.M.; Trojanowski, J.Q.; Fraser, N.W. Selective vulnerability of mouse CNS neurons to latent infection with a neuroattenuated herpes simplex virus-1. J. Neurosci. 1996, 16, 5644–5653. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kambara, H.; Okano, H.; Chiocca, E.A.; Saeki, Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005, 65, 2832–2839. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlstrand, J.; Collins, V.P.; Lendahl, U. Expression of the class VI intermediate filament nestin in human central nervous system tumors. Cancer Res. 1992, 52, 5334–5341. [Google Scholar] [PubMed]
- Kanai, R.; Tomita, H.; Hirose, Y.; Ohba, S.; Goldman, S.; Okano, H.; Kawase, T.; Yazaki, T. Augmented therapeutic efficacy of an oncolytic herpes simplex virus type 1 mutant expressing ICP34.5 under the transcriptional control of musashi1 promoter in the treatment of malignant glioma. Hum. Gene Ther. 2007, 18, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Hardcastle, J.; Kurozumi, K.; Chiocca, E.A.; Kaur, B. Oncolytic viruses driven by tumor-specific promoters. Curr. Cancer Drug Targets 2007, 7, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Marshall, E.E.; Bierle, C.J.; Brune, W.; Geballe, A.P. Essential role for either TRS1 or IRS1 in human cytomegalovirus replication. J. Virol. 2009, 83, 4112–4120. [Google Scholar] [CrossRef] [Green Version]
- Friedman, G.K.; Nan, L.; Haas, M.C.; Kelly, V.M.; Moore, B.P.; Langford, C.P.; Xu, H.; Han, X.; Beierle, E.A.; Markert, J.M.; et al. Gamma (1) 34.5-deleted HSV-1-expressing human cytomegalovirus IRS1 gene kills human glioblastoma cells as efficiently as wild-type HSV-1 in normoxia or hypoxia. Gene Ther. 2015, 22, 348–355. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Kimura, H.; Daikoku, T. Complementary lethal invasion of the central nervous system by nonneuroinvasive herpes simplex virus types 1 and 2. J. Virol. 1991, 65, 4520–4524. [Google Scholar] [CrossRef] [Green Version]
- Ushijima, Y.; Luo, C.; Goshima, F.; Yamauchi, Y.; Kimura, H.; Nishiyama, Y. Determination and analysis of the DNA sequence of highly attenuated herpes simplex virus type 1 mutant HF10, a potential oncolytic virus. Microbes Infect. 2007, 9, 142–149. [Google Scholar] [CrossRef]
- Alessandrini, F.; Ceresa, D.; Appolloni, I.; Marubbi, D.; Malatesta, P. Noninvasive Monitoring of Glioma Growth in the Mouse. J. Cancer 2016, 7, 1791–1797. [Google Scholar] [CrossRef] [Green Version]
- Sahin, T.T.; Kasuya, H.; Nomura, N.; Shikano, T.; Yamamura, K.; Gewen, T.; Kanzaki, A.; Fujii, T.; Sugae, T.; Imai, T.; et al. Impact of novel oncolytic virus HF10 on cellular components of the tumor microenviroment in patients with recurrent breast cancer. Cancer Gene Ther. 2012, 19, 229–237. [Google Scholar] [CrossRef] [PubMed]
- Takano, G.; Esaki, S.; Goshima, F.; Enomoto, A.; Hatano, Y.; Ozaki, H.; Watanabe, T.; Sato, Y.; Kawakita, D.; Murakami, S.; et al. Oncolytic activity of naturally attenuated herpes-simplex virus HF10 against an immunocompetent model of oral carcinoma. Mol. Ther. Oncolytics 2021, 20, 220–227. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.A.; Bates, P.; Rivera-Gonzalez, R.; Gu, B.; DeLuca, N.A. ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. J. Virol. 1993, 67, 4676–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piwecka, M.; Rolle, K.; Belter, A.; Barciszewska, A.M.; Zywicki, M.; Michalak, M.; Nowak, S.; Naskret-Barciszewska, M.Z.; Barciszewski, J. Comprehensive analysis of microRNA expression profile in malignant glioma tissues. Mol. Oncol. 2015, 9, 1324–1340. [Google Scholar] [CrossRef] [PubMed]
- Mazzacurati, L.; Marzulli, M.; Reinhart, B.; Miyagawa, Y.; Uchida, H.; Goins, W.F.; Li, A.; Kaur, B.; Caligiuri, M.; Cripe, T.; et al. Use of miRNA response sequences to block off-target replication and increase the safety of an unattenuated, glioblastoma-targeted oncolytic HSV. Mol. Ther. 2015, 23, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.Y.; Rennie, P.S.; Jia, W.W. MicroRNA regulation of oncolytic herpes simplex virus-1 for selective killing of prostate cancer cells. Clin. Cancer Res. 2009, 15, 5126–5135. [Google Scholar] [CrossRef] [Green Version]
- Kuroda, T.; Rabkin, S.D.; Martuza, R.L. Effective treatment of tumors with strong beta-catenin/T-cell factor activity by transcriptionally targeted oncolytic herpes simplex virus vector. Cancer Res. 2006, 66, 10127–10135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamamura, H.; Hashio, M.; Noguchi, M.; Sugenoya, Y.; Osakada, M.; Hirano, N.; Sasaki, Y.; Yoden, T.; Awata, N.; Araki, N.; et al. Identification of the transcriptional regulatory sequences of human calponin promoter and their use in targeting a conditionally replicating herpes vector to malignant human soft tissue and bone tumors. Cancer Res. 2001, 61, 3969–3977. [Google Scholar] [PubMed]
- Mineta, T.; Rabkin, S.D.; Martuza, R.L. Treatment of malignant gliomas using ganciclovir-hypersensitive, ribonucleotide reductase-deficient herpes simplex viral mutant. Cancer Res. 1994, 54, 3963–3966. [Google Scholar] [PubMed]
- Chase, M.; Chung, R.Y.; Chiocca, E.A. An oncolytic viral mutant that delivers the CYP2B1 transgene and augments cyclophosphamide chemotherapy. Nat. Biotechnol. 1998, 16, 444–448. [Google Scholar] [CrossRef] [PubMed]
- Mineta, T.; Rabkin, S.D.; Yazaki, T.; Hunter, W.D.; Martuza, R.L. Attenuated multi-mutated herpes simplex virus-1 for the treatment of malignant gliomas. Nat. Med. 1995, 1, 938–943. [Google Scholar] [CrossRef]
- Todo, T.; Martuza, R.L.; Rabkin, S.D.; Johnson, P.A. Oncolytic herpes simplex virus vector with enhanced MHC class I presentation and tumor cell killing. Proc. Natl. Acad. Sci. USA 2001, 98, 6396–6401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menotti, L.; Cerretani, A.; Hengel, H.; Campadelli-Fiume, G. Construction of a fully retargeted herpes simplex virus 1 recombinant capable of entering cells solely via human epidermal growth factor receptor 2. J. Virol. 2008, 82, 10153–10161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menotti, L.; Nicoletti, G.; Gatta, V.; Croci, S.; Landuzzi, L.; De Giovanni, C.; Nanni, P.; Lollini, P.L.; Campadelli-Fiume, G. Inhibition of human tumor growth in mice by an oncolytic herpes simplex virus designed to target solely HER-2-positive cells. Proc. Natl. Acad. Sci. USA 2009, 106, 9039–9044. [Google Scholar] [CrossRef] [Green Version]
- Nanni, P.; Gatta, V.; Menotti, L.; De Giovanni, C.; Ianzano, M.; Palladini, A.; Grosso, V.; Dall’ora, M.; Croci, S.; Nicoletti, G.; et al. Preclinical therapy of disseminated HER-2(+) ovarian and breast carcinomas with a HER-2-retargeted oncolytic herpesvirus. PLoS Pathog. 2013, 9, e1003155. [Google Scholar] [CrossRef] [Green Version]
- Reisoli, E.; Gambini, E.; Appolloni, I.; Gatta, V.; Barilari, M.; Menotti, L.; Malatesta, P. Efficacy of HER2 retargeted herpes simplex virus as therapy for high-grade glioma in immunocompetent mice. Cancer Gene Ther. 2012, 19, 788–795. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Vannini, A.; Gatta, V.; Rambaldi, J.; Sanapo, M.; Barboni, C.; Zaghini, A.; Nanni, P.; Lollini, P.L.; Casiraghi, C.; et al. A fully-virulent retargeted oncolytic HSV armed with IL-12 elicits local immunity and vaccine therapy towards distant tumors. PLoS Pathog. 2018, 14, e1007209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menotti, L.; Avitabile, E.; Gatta, V.; Malatesta, P.; Petrovic, B.; Campadelli-Fiume, G. HSV as A Platform for the Generation of Retargeted, Armed, and Reporter-Expressing Oncolytic Viruses. Viruses 2018, 10, 352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menotti, L.; Cerretani, A.; Campadelli-Fiume, G. A herpes simplex virus recombinant that exhibits a single-chain antibody to HER2/neu enters cells through the mammary tumor receptor, independently of the gD receptors. J. Virol. 2006, 80, 5531–5539. [Google Scholar] [CrossRef] [Green Version]
- Gambini, E.; Reisoli, E.; Appolloni, I.; Gatta, V.; Campadelli-Fiume, G.; Menotti, L.; Malatesta, P. Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol. Ther. 2012, 20, 994–1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchida, H.; Marzulli, M.; Nakano, K.; Goins, W.F.; Chan, J.; Hong, C.S.; Mazzacurati, L.; Yoo, J.Y.; Haseley, A.; Nakashima, H.; et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol. Ther. 2013, 21, 561–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powers, G.L.; Ellison-Zelski, S.J.; Casa, A.J.; Lee, A.V.; Alarid, E.T. Proteasome inhibition represses ERalpha gene expression in ER+ cells: A new link between proteasome activity and estrogen signaling in breast cancer. Oncogene 2010, 29, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, Y.; Zhang, J.; Wang, H.; Zhang, Y.; Sun, R.; Zhang, Z.; Gao, F.; Huang, C.; Zhang, S. Poorer prognosis in patients with advanced gastric squamous cell carcinoma compared with adenocarcinoma of the stomach: Case report. Medicine (Baltimore) 2017, 96, e9224. [Google Scholar] [CrossRef] [PubMed]
- Swaika, A.; Paulus, A.; Miller, K.C.; Sher, T.; Almyroudis, N.G.; Ball, D.; Wood, M.; Masood, A.; Lee, K.; Chanan-Khan, A.A. Acyclovir prophylaxis against varicella zoster virus reactivation in multiple myeloma patients treated with bortezomib-based therapies: A retrospective analysis of 100 patients. J. Supportive Oncol. 2012, 10, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Yoo, J.Y.; Jaime-Ramirez, A.C.; Bolyard, C.; Dai, H.; Nallanagulagari, T.; Wojton, J.; Hurwitz, B.S.; Relation, T.; Lee, T.J.; Lotze, M.T.; et al. Bortezomib Treatment Sensitizes Oncolytic HSV-1-Treated Tumors to NK Cell Immunotherapy. Clin. Cancer Res. 2016, 22, 5265–5276. [Google Scholar] [CrossRef] [Green Version]
- Pannuti, A.; Foreman, K.; Rizzo, P.; Osipo, C.; Golde, T.; Osborne, B.; Miele, L. Targeting Notch to target cancer stem cells. Clin. Cancer Res. 2010, 16, 3141–3152. [Google Scholar] [CrossRef] [Green Version]
- Otani, Y.; Yoo, J.Y.; Chao, S.; Liu, J.; Jaime-Ramirez, A.C.; Lee, T.J.; Hurwitz, B.; Yan, Y.; Dai, H.; Glorioso, J.C.; et al. Oncolytic HSV-Infected Glioma Cells Activate NOTCH in Adjacent Tumor Cells Sensitizing Tumors to Gamma Secretase Inhibition. Clin. Cancer Res. 2020, 26, 2381–2392. [Google Scholar] [CrossRef] [Green Version]
- Jennings, V.A.; Scott, G.B.; Rose, A.M.S.; Scott, K.J.; Migneco, G.; Keller, B.; Reilly, K.; Donnelly, O.; Peach, H.; Dewar, D.; et al. Potentiating Oncolytic Virus-Induced Immune-Mediated Tumor Cell Killing Using Histone Deacetylase Inhibition. Mol. Ther. 2019, 27, 1139–1152. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, H.; Kaufmann, J.K.; Wang, P.Y.; Nguyen, T.; Speranza, M.C.; Kasai, K.; Okemoto, K.; Otsuki, A.; Nakano, I.; Fernandez, S.; et al. Histone deacetylase 6 inhibition enhances oncolytic viral replication in glioma. J. Clin. Investig. 2015, 125, 4269–4280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommareddy, P.K.; Zloza, A.; Rabkin, S.D.; Kaufman, H.L. Oncolytic virus immunotherapy induces immunogenic cell death and overcomes STING deficiency in melanoma. Oncoimmunology 2019, 8, 1591875. [Google Scholar] [CrossRef]
- Ishino, R.; Kawase, Y.; Kitawaki, T.; Sugimoto, N.; Oku, M.; Uchida, S.; Imataki, O.; Matsuoka, A.; Taoka, T.; Kawakami, K.; et al. Oncolytic Virus Therapy with HSV-1 for Hematological Malignancies. Mol. Ther. 2021, 29, 762–774. [Google Scholar] [CrossRef] [PubMed]
- Woo, S.R.; Fuertes, M.B.; Corrales, L.; Spranger, S.; Furdyna, M.J.; Leung, M.Y.; Duggan, R.; Wang, Y.; Barber, G.N.; Fitzgerald, K.A.; et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014, 41, 830–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Froechlich, G.; Caiazza, C.; Gentile, C.; D’Alise, A.M.; De Lucia, M.; Langone, F.; Leoni, G.; Cotugno, G.; Scisciola, V.; Nicosia, A.; et al. Integrity of the Antiviral STING-mediated DNA Sensing in Tumor Cells Is Required to Sustain the Immunotherapeutic Efficacy of Herpes Simplex Oncolytic Virus. Cancers 2020, 12, 3407. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; Chapa, V.; Saini, U.; Modgil, P.; Cohn, D.E.; He, G.; Siddik, Z.H.; Sood, A.K.; Yan, Y.; Selvendiran, K.; et al. Oncolytic HSV Therapy Modulates Vesicular Trafficking Inducing Cisplatin Sensitivity and Antitumor Immunity. Clin. Cancer Res. 2021, 27, 542–553. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y. Temozolomide resistance in glioblastoma multiforme. Genes Dis. 2016, 3, 198–210. [Google Scholar] [CrossRef] [Green Version]
- Kanai, R.; Rabkin, S.D.; Yip, S.; Sgubin, D.; Zaupa, C.M.; Hirose, Y.; Louis, D.N.; Wakimoto, H.; Martuza, R.L. Oncolytic virus-mediated manipulation of DNA damage responses: Synergy with chemotherapy in killing glioblastoma stem cells. J. Natl Cancer Inst. 2012, 104, 42–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mezhir, J.J.; Advani, S.J.; Smith, K.D.; Darga, T.E.; Poon, A.P.; Schmidt, H.; Posner, M.C.; Roizman, B.; Weichselbaum, R.R. Ionizing radiation activates late herpes simplex virus 1 promoters via the p38 pathway in tumors treated with oncolytic viruses. Cancer Res. 2005, 65, 9479–9484. [Google Scholar] [CrossRef] [Green Version]
- Markert, J.M.; Razdan, S.N.; Kuo, H.C.; Cantor, A.; Knoll, A.; Karrasch, M.; Nabors, L.B.; Markiewicz, M.; Agee, B.S.; Coleman, J.M.; et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol. Ther. 2014, 22, 1048–1055. [Google Scholar] [CrossRef] [Green Version]
- Jarnagin, W.R.; Zager, J.S.; Hezel, M.; Stanziale, S.F.; Adusumilli, P.S.; Gonen, M.; Ebright, M.I.; Culliford, A.; Gusani, N.J.; Fong, Y. Treatment of cholangiocarcinoma with oncolytic herpes simplex virus combined with external beam radiation therapy. Cancer Gene Ther. 2006, 13, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Long, G.V.; Stroyakovskiy, D.; Gogas, H.; Levchenko, E.; de Braud, F.; Larkin, J.; Garbe, C.; Jouary, T.; Hauschild, A.; Grob, J.J.; et al. Combined BRAF and MEK inhibition versus BRAF inhibition alone in melanoma. N. Engl. J. Med. 2014, 371, 1877–1888. [Google Scholar] [CrossRef] [Green Version]
- Ebert, P.J.R.; Cheung, J.; Yang, Y.; McNamara, E.; Hong, R.; Moskalenko, M.; Gould, S.E.; Maecker, H.; Irving, B.A.; Kim, J.M.; et al. MAP Kinase Inhibition Promotes T Cell and Anti-tumor Activity in Combination with PD-L1 Checkpoint Blockade. Immunity 2016, 44, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Smith, K.D.; Mezhir, J.J.; Bickenbach, K.; Veerapong, J.; Charron, J.; Posner, M.C.; Roizman, B.; Weichselbaum, R.R. Activated MEK suppresses activation of PKR and enables efficient replication and in vivo oncolysis by Deltagamma(1)34.5 mutants of herpes simplex virus 1. J. Virol. 2006, 80, 1110–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu-Lieskovan, S.; Robert, L.; Homet Moreno, B.; Ribas, A. Combining targeted therapy with immunotherapy in BRAF-mutant melanoma: Promise and challenges. J. Clin. Oncol. 2014, 32, 2248–2254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bommareddy, P.K.; Aspromonte, S.; Zloza, A.; Rabkin, S.D.; Kaufman, H.L. MEK inhibition enhances oncolytic virus immunotherapy through increased tumor cell killing and T cell activation. Sci. Transl. Med. 2018, 10, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoo, J.Y.; Swanner, J.; Otani, Y.; Nair, M.; Park, F.; Banasavadi-Siddegowda, Y.; Liu, J.; Jaime-Ramirez, A.C.; Hong, B.; Geng, F.; et al. Oncolytic HSV therapy increases trametinib access to brain tumors and sensitizes them in vivo. Neuro-Oncology 2019, 21, 1131–1140. [Google Scholar] [CrossRef]
- Liu, T.C.; Wakimoto, H.; Martuza, R.L.; Rabkin, S.D. Herpes simplex virus Us3(−) mutant as oncolytic strategy and synergizes with phosphatidylinositol 3-kinase-Akt targeting molecular therapeutics. Clin. Cancer Res. 2007, 13, 5897–5902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, R.; Wakimoto, H.; Martuza, R.L.; Rabkin, S.D. A novel oncolytic herpes simplex virus that synergizes with phosphoinositide 3-kinase/Akt pathway inhibitors to target glioblastoma stem cells. Clin. Cancer Res. 2011, 17, 3686–3696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, L.; Swanner, J.; Jaime-Ramirez, A.C.; Wang, Y.; Sprague, A.; Banasavadi-Siddegowda, Y.; Yoo, J.Y.; Sizemore, G.M.; Kladney, R.; Zhang, J.; et al. PTEN expression by an oncolytic herpesvirus directs T-cell mediated tumor clearance. Nat. Commun. 2018, 9, 5006. [Google Scholar] [CrossRef]
- Gatson, N.N.; Chiocca, E.A.; Kaur, B. Anti-angiogenic gene therapy in the treatment of malignant gliomas. Neurosci. Lett. 2012, 527, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Tan, G.; Kasuya, H.; Sahin, T.T.; Yamamura, K.; Wu, Z.; Koide, Y.; Hotta, Y.; Shikano, T.; Yamada, S.; Kanzaki, A.; et al. Combination therapy of oncolytic herpes simplex virus HF10 and bevacizumab against experimental model of human breast carcinoma xenograft. Int. J. Cancer 2015, 136, 1718–1730. [Google Scholar] [CrossRef] [PubMed]
- Kurozumi, K.; Hardcastle, J.; Thakur, R.; Yang, M.; Christoforidis, G.; Fulci, G.; Hochberg, F.H.; Weissleder, R.; Carson, W.; Chiocca, E.A.; et al. Effect of tumor microenvironment modulation on the efficacy of oncolytic virus therapy. J. Natl. Cancer Inst. 2007, 99, 1768–1781. [Google Scholar] [CrossRef] [Green Version]
- Nair, M.; Khosla, M.; Otani, Y.; Yeh, M.; Park, F.; Shimizu, T.; Kang, J.M.; Bolyard, C.; Yu, J.G.; Kumar Banasavadi-Siddegowda, Y.; et al. Enhancing Antitumor Efficacy of Heavily Vascularized Tumors by RAMBO Virus through Decreased Tumor Endothelial Cell Activation. Cancers 2020, 12, 1040. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Pradarelli, J.; Haseley, A.; Wojton, J.; Kaka, A.; Bratasz, A.; Alvarez-Breckenridge, C.A.; Yu, J.G.; Powell, K.; Mazar, A.P.; et al. Copper chelation enhances antitumor efficacy and systemic delivery of oncolytic HSV. Clin. Cancer Res. 2012, 18, 4931–4941. [Google Scholar] [CrossRef] [Green Version]
- Alvarez-Breckenridge, C.A.; Yu, J.; Price, R.; Wojton, J.; Pradarelli, J.; Mao, H.; Wei, M.; Wang, Y.; He, S.; Hardcastle, J.; et al. NK cells impede glioblastoma virotherapy through NKp30 and NKp46 natural cytotoxicity receptors. Nat. Med. 2012, 18, 1827–1834. [Google Scholar] [CrossRef] [Green Version]
- Dai, H.S.; Griffin, N.; Bolyard, C.; Mao, H.C.; Zhang, J.; Cripe, T.P.; Suenaga, T.; Arase, H.; Nakano, I.; Chiocca, E.A.; et al. The Fc Domain of Immunoglobulin Is Sufficient to Bridge NK Cells with Virally Infected Cells. Immunity 2017, 47, 159–170.e10. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Han, J.; Chu, J.; Zhang, L.; Zhang, J.; Chen, C.; Chen, L.; Wang, Y.; Wang, H.; Yi, L.; et al. A combinational therapy of EGFR-CAR NK cells and oncolytic herpes simplex virus 1 for breast cancer brain metastases. Oncotarget 2016, 7, 27764–27777. [Google Scholar] [CrossRef] [Green Version]
- Han, J.; Chen, X.; Chu, J.; Xu, B.; Meisen, W.H.; Chen, L.; Zhang, L.; Zhang, J.; He, X.; Wang, Q.E.; et al. TGFbeta Treatment Enhances Glioblastoma Virotherapy by Inhibiting the Innate Immune Response. Cancer Res. 2015, 75, 5273–5282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Yoo, J.Y.; Lee, T.J.; Liu, J.; Yu, J.; Caligiuri, M.A.; Kaur, B.; Friedman, A. Complex role of NK cells in regulation of oncolytic virus-bortezomib therapy. Proc. Natl. Acad. Sci. USA 2018, 115, 4927–4932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorne, A.H.; Meisen, W.H.; Russell, L.; Yoo, J.Y.; Bolyard, C.M.; Lathia, J.D.; Rich, J.; Puduvalli, V.K.; Mao, H.; Yu, J.; et al. Role of cysteine-rich 61 protein (CCN1) in macrophage-mediated oncolytic herpes simplex virus clearance. Mol. Ther. 2014, 22, 1678–1687. [Google Scholar] [CrossRef] [Green Version]
- Delwar, Z.M.; Kuo, Y.; Wen, Y.H.; Rennie, P.S.; Jia, W. Oncolytic Virotherapy Blockade by Microglia and Macrophages Requires STAT1/3. Cancer Res. 2018, 78, 718–730. [Google Scholar] [CrossRef] [Green Version]
- Bolyard, C.; Meisen, W.H.; Banasavadi-Siddegowda, Y.; Hardcastle, J.; Yoo, J.Y.; Wohleb, E.S.; Wojton, J.; Yu, J.G.; Dubin, S.; Khosla, M.; et al. BAI1 Orchestrates Macrophage Inflammatory Response to HSV Infection-Implications for Oncolytic Viral Therapy. Clin. Cancer Res. 2017, 23, 1809–1819. [Google Scholar] [CrossRef] [Green Version]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.S.; Yu, J.; Kaur, B.; Louis, D.N.; Weissleder, R.; Caligiuri, M.A.; et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, N.L.; Chen, C.Y.; Hutzen, B.; Currier, M.A.; Scott, T.; Nartker, B.; Leddon, J.L.; Wang, P.Y.; Srinivas, R.; Cassady, K.A.; et al. Myelolytic Treatments Enhance Oncolytic Herpes Virotherapy in Models of Ewing Sarcoma by Modulating the Immune Microenvironment. Mol. Ther. Oncolytics 2018, 11, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, K.; Wakimoto, H.; Ichikawa, T.; Jhung, S.; Hochberg, F.H.; Louis, D.N.; Chiocca, E.A. Complement depletion facilitates the infection of multiple brain tumors by an intravascular, replication-conditional herpes simplex virus mutant. J. Virol. 2000, 74, 4765–4775. [Google Scholar] [CrossRef] [Green Version]
- Yoo, J.Y.; Yu, J.G.; Kaka, A.; Pan, Q.; Kumar, P.; Kumar, B.; Zhang, J.; Mazar, A.; Teknos, T.N.; Kaur, B.; et al. ATN-224 enhances antitumor efficacy of oncolytic herpes virus against both local and metastatic head and neck squamous cell carcinoma. Mol. Ther. Oncolytics 2015, 2, 15008. [Google Scholar] [CrossRef] [PubMed]
- Wakimoto, H.; Johnson, P.R.; Knipe, D.M.; Chiocca, E.A. Effects of innate immunity on herpes simplex virus and its ability to kill tumor cells. Gene Ther. 2003, 10, 983–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, K.; Ichikawa, T.; Wakimoto, H.; Silver, J.S.; Deisboeck, T.S.; Finkelstein, D.; Harsh, G.R.t.; Louis, D.N.; Bartus, R.T.; Hochberg, F.H.; et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat. Med. 1999, 5, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Parker Kerrigan, B.C.; Shimizu, Y.; Andreeff, M.; Lang, F.F. Mesenchymal stromal cells for the delivery of oncolytic viruses in gliomas. Cytotherapy 2017, 19, 445–457. [Google Scholar] [CrossRef] [Green Version]
- Duebgen, M.; Martinez-Quintanilla, J.; Tamura, K.; Hingtgen, S.; Redjal, N.; Wakimoto, H.; Shah, K. Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J. Natl. Cancer Inst. 2014, 106, dju090. [Google Scholar] [CrossRef]
- Krause, M.N.; Sancho-Martinez, I.; Izpisua Belmonte, J.C. RE: Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J. Natl. Cancer Inst. 2015, 107, 368. [Google Scholar] [CrossRef] [Green Version]
- Israyelyan, A.H.; Melancon, J.M.; Lomax, L.G.; Sehgal, I.; Leuschner, C.; Kearney, M.T.; Chouljenko, V.N.; Baghian, A.; Kousoulas, K.G. Effective treatment of human breast tumor in a mouse xenograft model with herpes simplex virus type 1 specifying the NV1020 genomic deletion and the gBsyn3 syncytial mutation enabling high viral replication and spread in breast cancer cells. Hum. Gene Ther. 2007, 18, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Uchida, H.; Shibata, T.; Sasaki, Y.; Ikeda, H.; Hamada-Uematsu, M.; Hamasaki, R.; Okuda, K.; Yanagi, S.; Tahara, H. Potent anti-tumor effects of receptor-retargeted syncytial oncolytic herpes simplex virus. Mol. Ther. Oncolytics 2021, 22, 265–276. [Google Scholar] [CrossRef]
- Herrlinger, U.; Woiciechowski, C.; Sena-Esteves, M.; Aboody, K.S.; Jacobs, A.H.; Rainov, N.G.; Snyder, E.Y.; Breakefield, X.O. Neural precursor cells for delivery of replication-conditional HSV-1 vectors to intracerebral gliomas. Mol. Ther. 2000, 1, 347–357. [Google Scholar] [CrossRef]
- Kanzaki, A.; Kasuya, H.; Yamamura, K.; Sahin, T.T.; Nomura, N.; Shikano, T.; Shirota, T.; Tan, G.; Fukuda, S.; Misawa, M.; et al. Antitumor efficacy of oncolytic herpes simplex virus adsorbed onto antigen-specific lymphocytes. Cancer Gene Ther. 2012, 19, 292–298. [Google Scholar] [CrossRef] [Green Version]
- McKee, T.D.; Grandi, P.; Mok, W.; Alexandrakis, G.; Insin, N.; Zimmer, J.P.; Bawendi, M.G.; Boucher, Y.; Breakefield, X.O.; Jain, R.K. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006, 66, 2509–2513. [Google Scholar] [CrossRef] [Green Version]
- Shintani, M.; Takahashi, G.; Hamada, M.; Okunaga, S.; Iwai, S.; Yura, Y. Effect of ultrasound on herpes simplex virus infection in cell culture. Virol. J. 2011, 8, 446. [Google Scholar] [CrossRef] [Green Version]
- Fujii, K.; Kurozumi, K.; Ichikawa, T.; Onishi, M.; Shimazu, Y.; Ishida, J.; Chiocca, E.A.; Kaur, B.; Date, I. The integrin inhibitor cilengitide enhances the anti-glioma efficacy of vasculostatin-expressing oncolytic virus. Cancer Gene Ther. 2013, 20, 437–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, B.; Muili, K.; Bolyard, C.; Russell, L.; Lee, T.J.; Banasavadi-Siddegowda, Y.; Yoo, J.Y.; Yan, Y.; Ballester, L.Y.; Bockhorst, K.H.; et al. Suppression of HMGB1 Released in the Glioblastoma Tumor Microenvironment Reduces Tumoral Edema. Mol. Ther. Oncolytics 2019, 12, 93–102. [Google Scholar] [CrossRef] [Green Version]
- Eshun, F.K.; Currier, M.A.; Gillespie, R.A.; Fitzpatrick, J.L.; Baird, W.H.; Cripe, T.P. VEGF blockade decreases the tumor uptake of systemic oncolytic herpes virus but enhances therapeutic efficacy when given after virotherapy. Gene Ther. 2010, 17, 922–929. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Nace, R.; Ayala-Breton, C.C.; Steele, M.; Bailey, K.; Peng, K.W.; Russell, S.J. Perfusion Pressure Is a Critical Determinant of the Intratumoral Extravasation of Oncolytic Viruses. Mol. Ther. 2016, 24, 306–317. [Google Scholar] [CrossRef] [Green Version]
- Seymour, L.W.; Fisher, K.D. Under Pressure: Elevated Blood Pressure Enhances Targeting of Tumors by Oncolytic Viruses. Mol. Ther. 2016, 24, 204–205. [Google Scholar] [CrossRef] [Green Version]
- Sette, P.; Amankulor, N.; Li, A.; Marzulli, M.; Leronni, D.; Zhang, M.; Goins, W.F.; Kaur, B.; Bolyard, C.; Cripe, T.P.; et al. GBM-Targeted oHSV Armed with Matrix Metalloproteinase 9 Enhances Anti-tumor Activity and Animal Survival. Mol. Ther. Oncolytics 2019, 15, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.S.; Fellows, W.; Niranjan, A.; Alber, S.; Watkins, S.; Cohen, J.B.; Glorioso, J.C.; Grandi, P. Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Ther. 2010, 17, 1200–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedan, S.; Rojas, J.J.; Gros, A.; Mercade, E.; Cascallo, M.; Alemany, R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol. Ther. 2010, 18, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lee, H.G.; Dmitrieva, N.; Kim, J.; Kaur, B.; Friedman, A. Choindroitinase ABC I-mediated enhancement of oncolytic virus spread and anti tumor efficacy: A mathematical model. PLoS ONE 2014, 9, e102499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.H.; Lee, Y.S.; Kim, H.; Huang, J.H.; Yoon, A.R.; Yun, C.O. Relaxin expression from tumor-targeting adenoviruses and its intratumoral spread, apoptosis induction, and efficacy. J. Natl. Cancer Inst. 2006, 98, 1482–1493. [Google Scholar] [CrossRef]
- Jaime-Ramirez, A.C.; Dmitrieva, N.; Yoo, J.Y.; Banasavadi-Siddegowda, Y.; Zhang, J.; Relation, T.; Bolyard, C.; Wojton, J.; Kaur, B. Humanized chondroitinase ABC sensitizes glioblastoma cells to temozolomide. J. Gene Med. 2017, 19, e2942. [Google Scholar] [CrossRef] [Green Version]
- Dmitrieva, N.; Yu, L.; Viapiano, M.; Cripe, T.P.; Chiocca, E.A.; Glorioso, J.C.; Kaur, B. Chondroitinase ABC I-mediated enhancement of oncolytic virus spread and antitumor efficacy. Clin. Cancer Res. 2011, 17, 1362–1372. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.G.; Choi, Y.H.; Hong, J.; Choi, J.W.; Yoon, A.R.; Yun, C.O. GM101 in Combination with Histone Deacetylase Inhibitor Enhances Anti-Tumor Effects in Desmoplastic Microenvironment. Cells 2021, 10, 2811. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, B.; Sahu, U.; Mullarkey, M.P.; Kaur, B. Replication and Spread of Oncolytic Herpes Simplex Virus in Solid Tumors. Viruses 2022, 14, 118. https://doi.org/10.3390/v14010118
Hong B, Sahu U, Mullarkey MP, Kaur B. Replication and Spread of Oncolytic Herpes Simplex Virus in Solid Tumors. Viruses. 2022; 14(1):118. https://doi.org/10.3390/v14010118
Chicago/Turabian StyleHong, Bangxing, Upasana Sahu, Matthew P. Mullarkey, and Balveen Kaur. 2022. "Replication and Spread of Oncolytic Herpes Simplex Virus in Solid Tumors" Viruses 14, no. 1: 118. https://doi.org/10.3390/v14010118
APA StyleHong, B., Sahu, U., Mullarkey, M. P., & Kaur, B. (2022). Replication and Spread of Oncolytic Herpes Simplex Virus in Solid Tumors. Viruses, 14(1), 118. https://doi.org/10.3390/v14010118