

Design of Three Residues Peptides against SARS-CoV-2 Infection

 , ,
, ,  ,
,  ,
,  , ,
, ,  ,
,  ,
,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Alignment
2.2. Peptides Synthesis
2.3. Peptide Stability
2.4. Cells and Viruses
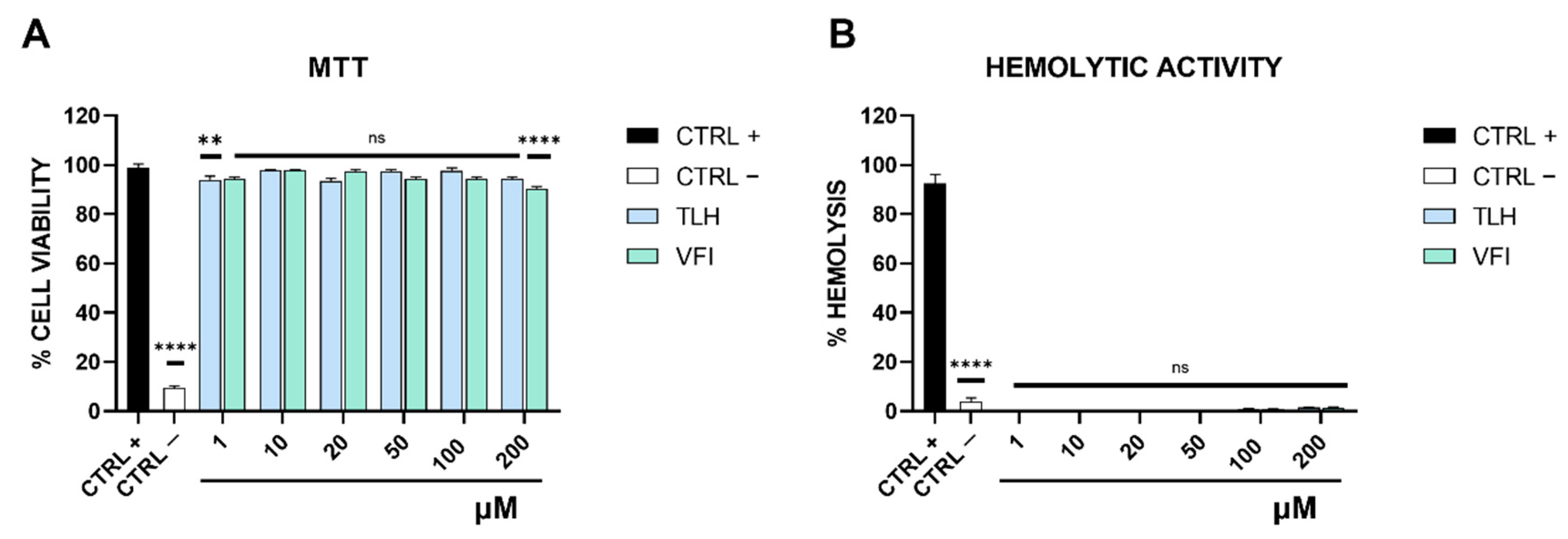
2.5. Cytotoxicity Assay
2.6. Hemolysis Assay
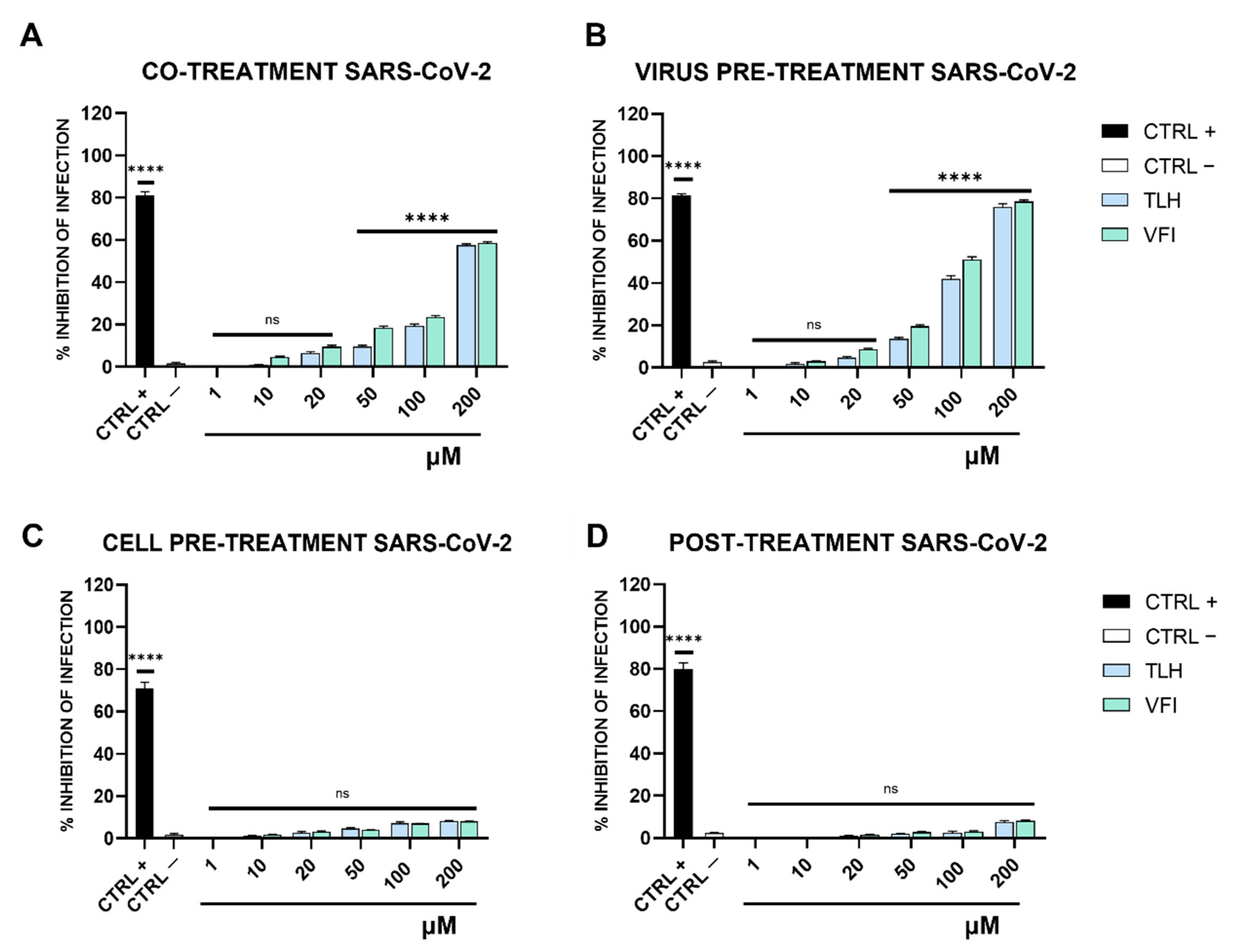
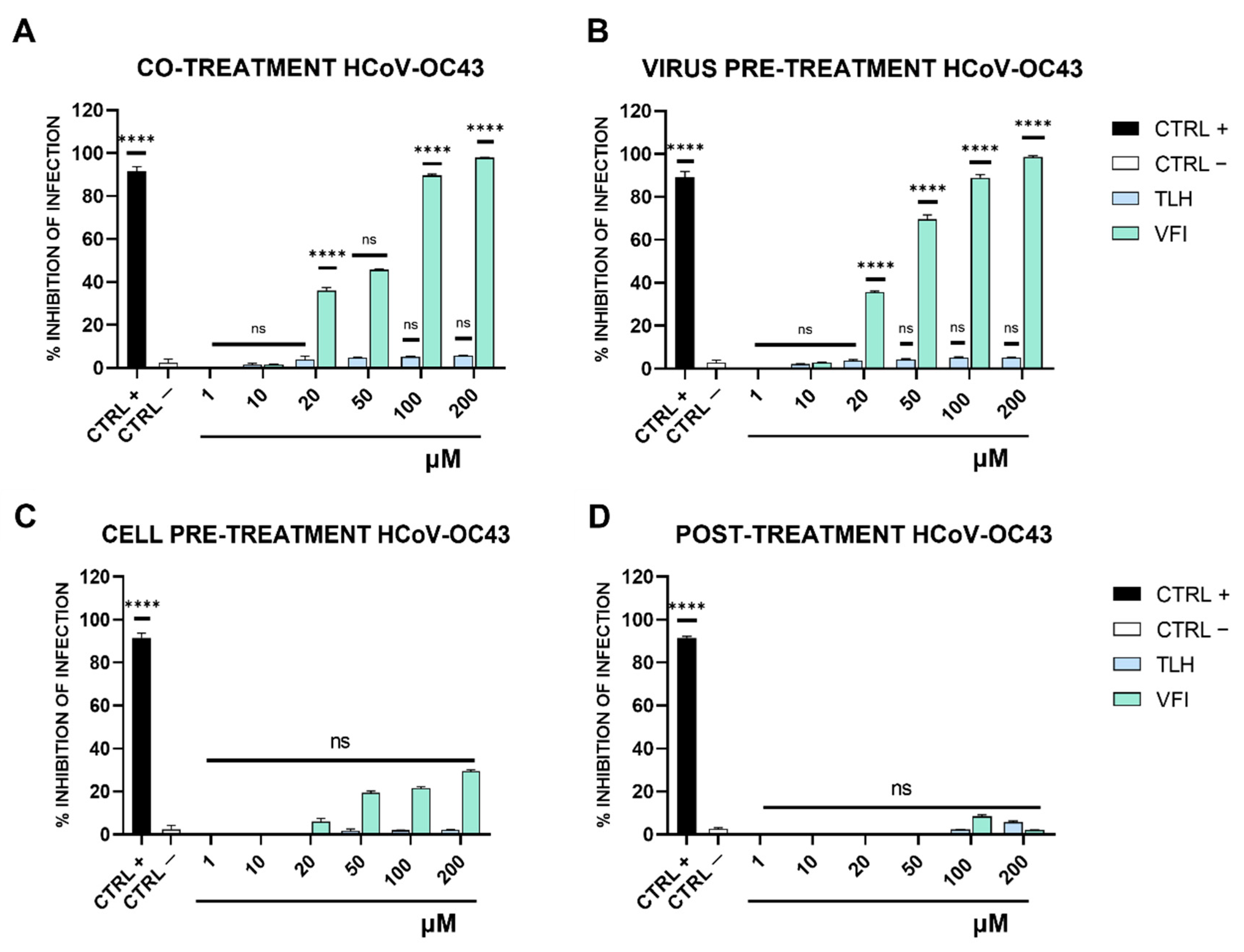
2.7. Antiviral Testing
- (a)
- Co-treatment test. This is a screening assay to point out the tripeptides’ activity as antiviral agents. Then, each tripeptide was added to the cell monolayer (1–200 μM) at the same time as viral infection at a multiplicity of infection (MOI) of 0.1 plaque forming unit (pfu)/cell for 2 h at 37 °C.
- (b)
- Virus pre-treatment. This test is useful for evaluating whether tripeptides can act directly on the viral particles. Each peptide was added to the virus (1 × 104 pfu/mL) and incubated for 1 h at 37 °C. After incubation, the mixture (virus/peptide) was diluted on cells and incubated for 2 supplementary hours, so that the peptide reaches a non-active concentration and the virus was at a MOI of 0.01 pfu/cell.
- (c)
- Cell pre-treatment. To assess whether tripeptides could interact with the target cell, preventing the subsequent binding to the virus surface. Cells were pre-cooled at 4 °C for 30 min and, subsequently, the peptide was added and incubated for 1 h at 4 °C. Then virus was added to a MOI of 0.1 pfu/mL for 2 h at 37 °C.
- (d)
- Post-treatment. The assay allows the assessment of the tripeptides’ ability to interfere with the viral replication. Cells were incubated with virus (MOI 0.1 pfu/mL) for 2 h at 37 °C, after that the peptide was added and incubated on the cells for two different time lapses, i.e., 1 h and 24 h.
2.8. Statistical Analysis
2.9. Molecular Docking
2.10. Octet Biolayer Interferometry (BLI)
3. Results
3.1. Design of Tripeptides
3.2. Peptides Toxicity
3.3. Antiviral Activity
3.4. In Silico Analysis
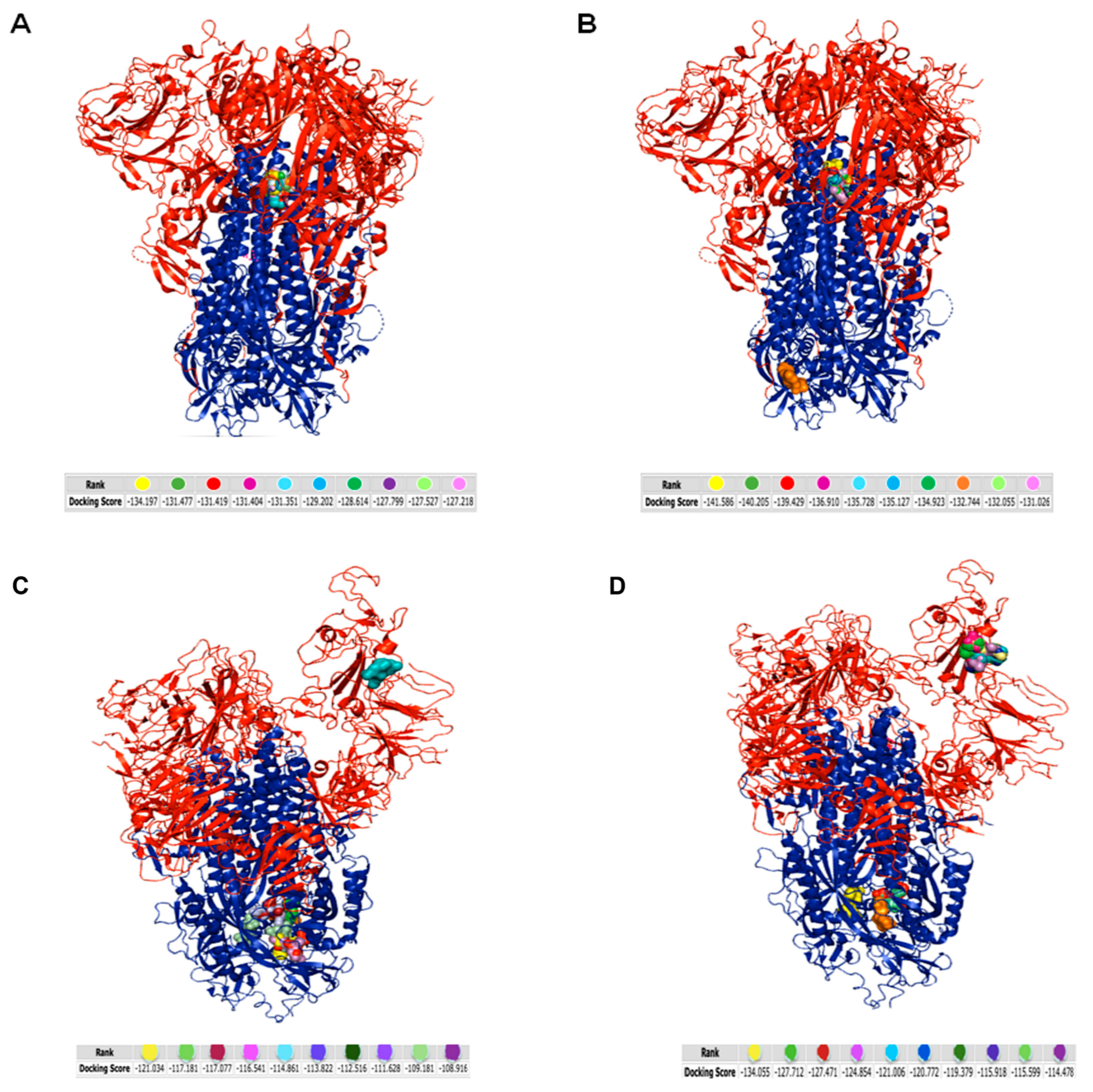
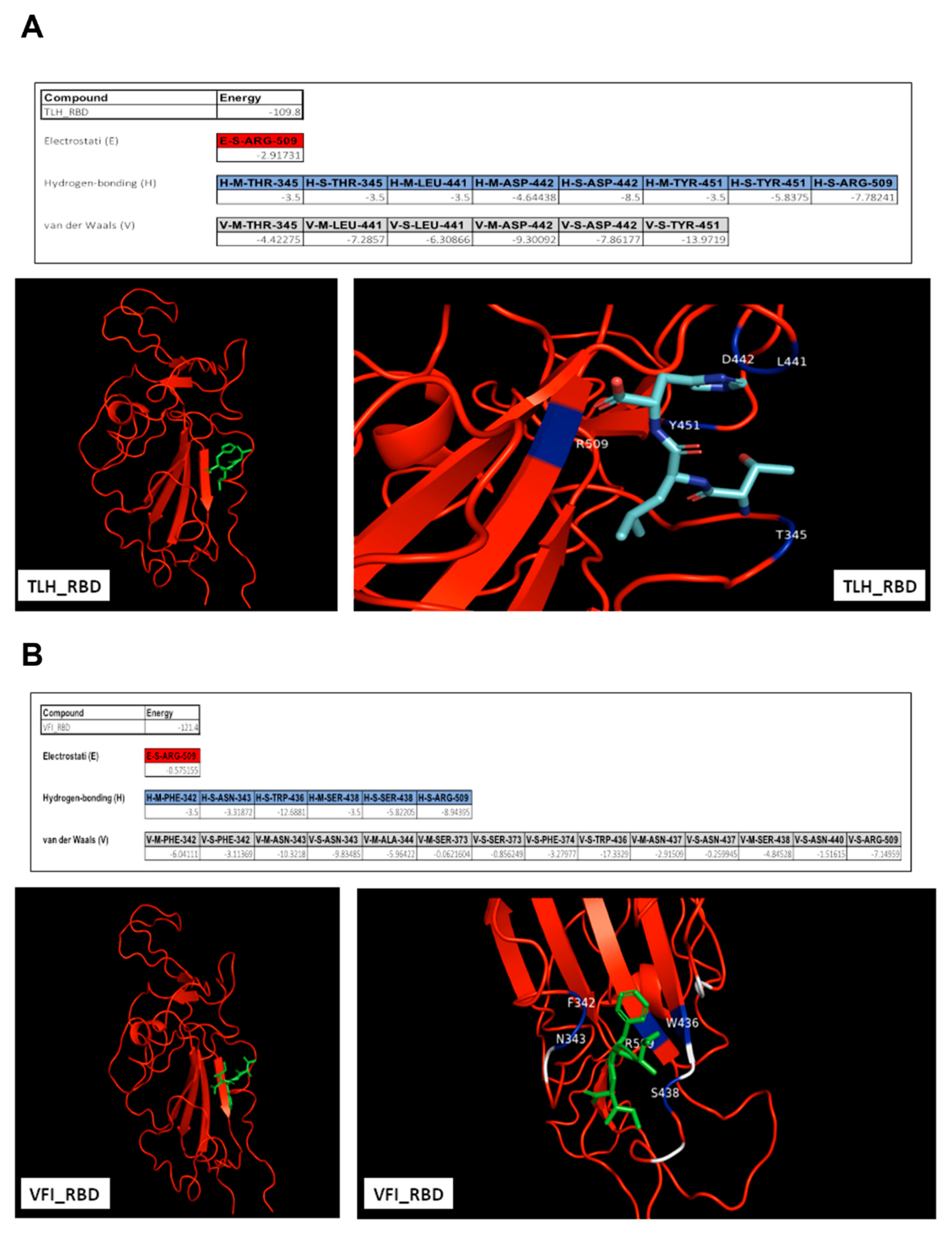
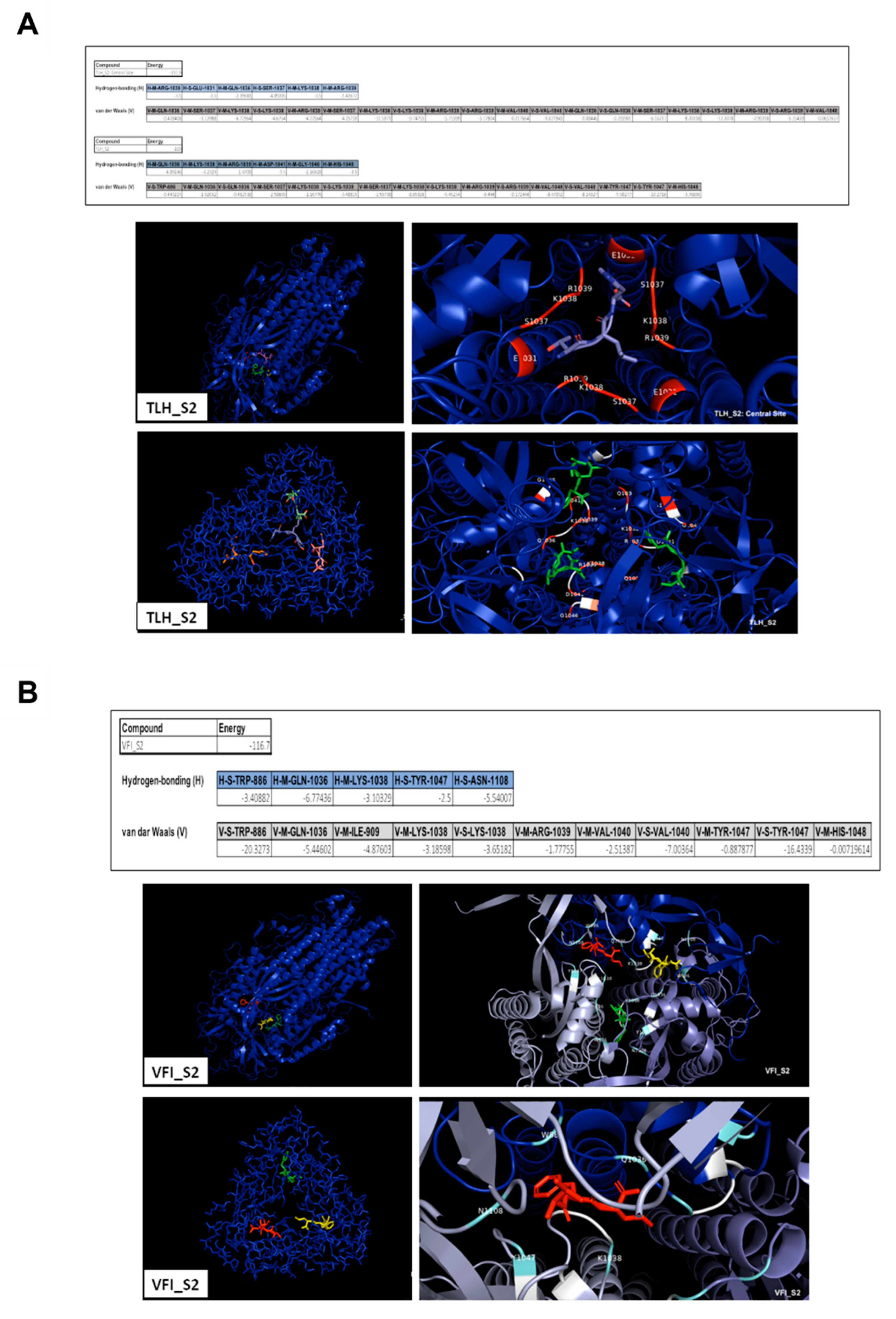
3.5. Analysis of the Interaction between Tripeptides and SARS-CoV-2 Spike Protein
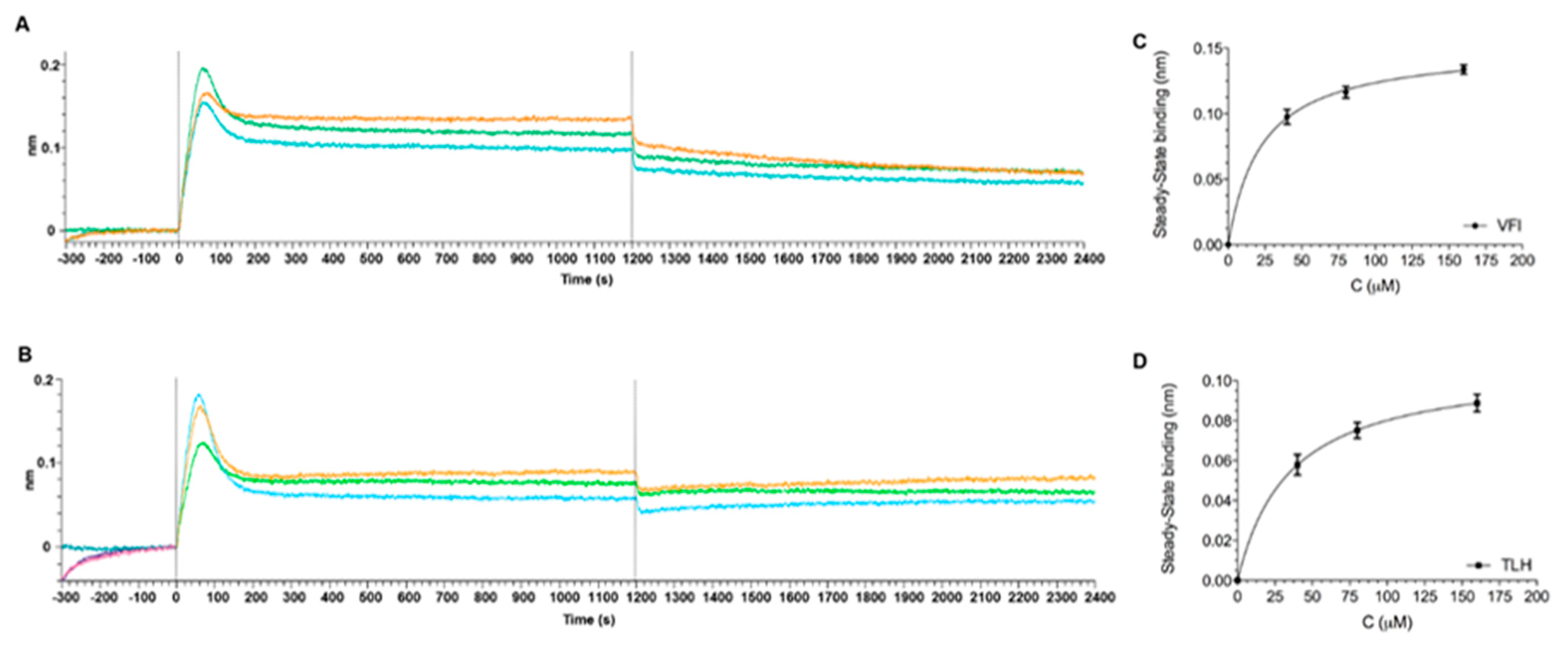
3.6. Direct Binding Assays between Tripeptides and SARS-CoV-2 Spike Protein
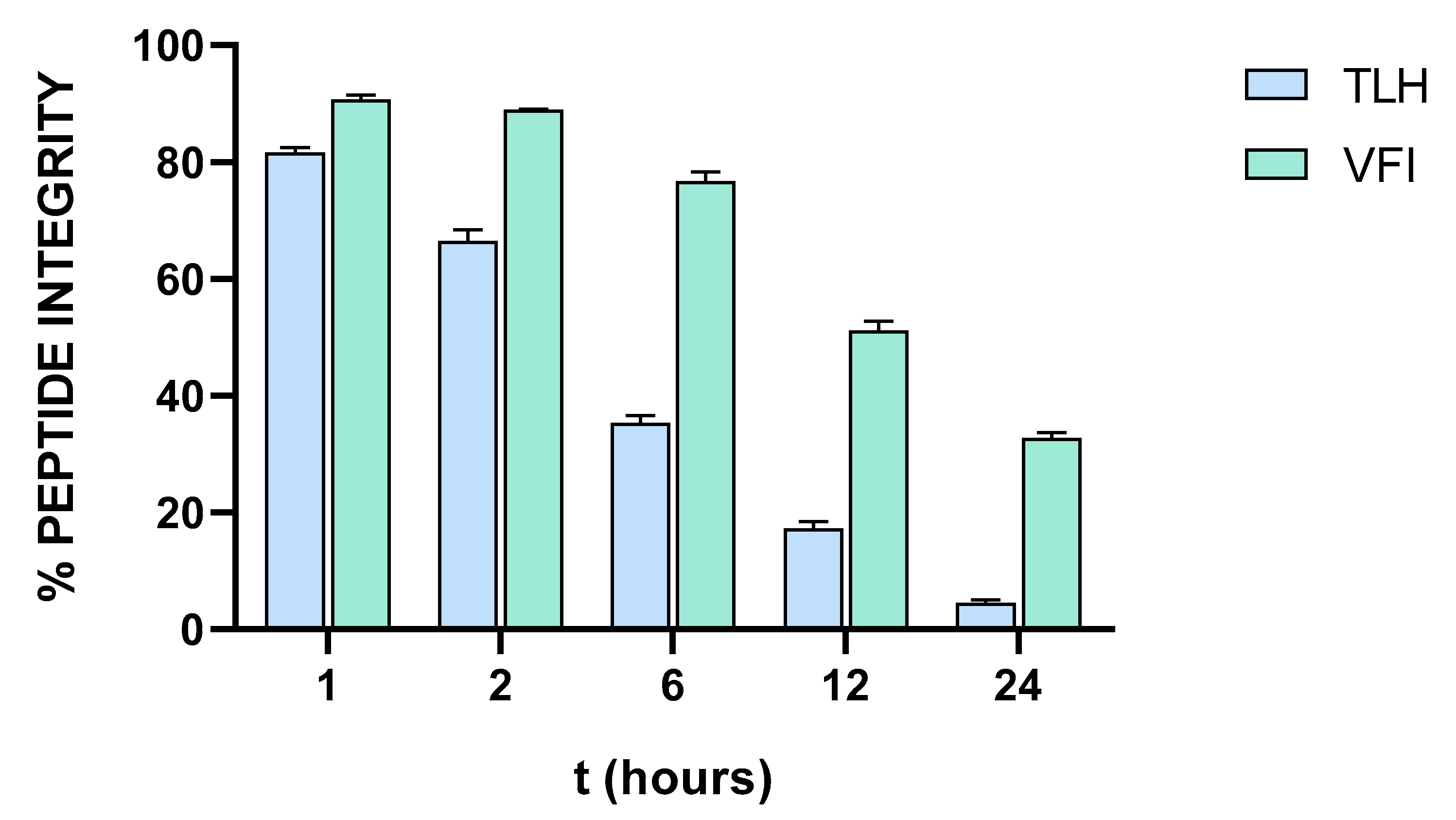
3.7. Stability of Tripeptides in Human Serum
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Galdiero, M.; Galdiero, M.; Folliero, V.; Zannella, C.; De Filippis, A.; Mali, A.; Rinaldi, L.; Franci, G. SARS-CoV-2 vaccine development: Where are we? Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 2752–2784. [Google Scholar] [PubMed]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Preston, D.J. Uganda safari (American surgeon visits east Africa). Del. Med. J. 1989, 61, 217–218, 222–223. [Google Scholar] [PubMed]
- Othman, H.; Bouslama, Z.; Brandenburg, J.T.; da Rocha, J.; Hamdi, Y.; Ghedira, K.; Srairi-Abid, N.; Hazelhurst, S. Interaction of the spike protein RBD from SARS-CoV-2 with ACE2: Similarity with SARS-CoV, hot-spot analysis and effect of the receptor polymorphism. Biochem. Biophys. Res. Commun. 2020, 527, 702–708. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef]
- Belouzard, S.; Millet, J.K.; Licitra, B.N.; Whittaker, G.R. Mechanisms of coronavirus cell entry mediated by the viral spike protein. Viruses 2012, 4, 1011–1033. [Google Scholar] [CrossRef]
- Fuentes-Prior, P. Priming of SARS-CoV-2 S protein by several membrane-bound serine proteinases could explain enhanced viral infectivity and systemic COVID-19 infection. J. Biol. Chem. 2021, 296, 100135. [Google Scholar] [CrossRef]
- Heurich, A.; Hofmann-Winkler, H.; Gierer, S.; Liepold, T.; Jahn, O.; Pohlmann, S. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2014, 88, 1293–1307. [Google Scholar] [CrossRef]
- Zhang, J.; Xiao, T.; Cai, Y.; Chen, B. Structure of SARS-CoV-2 spike protein. Curr. Opin. Virol. 2021, 50, 173–182. [Google Scholar] [CrossRef]
- Schutz, D.; Ruiz-Blanco, Y.B.; Munch, J.; Kirchhoff, F.; Sanchez-Garcia, E.; Muller, J.A. Peptide and peptide-based inhibitors of SARS-CoV-2 entry. Adv. Drug Deliv. Rev. 2020, 167, 47–65. [Google Scholar] [CrossRef]
- Mazzon, M.; Marsh, M. Targeting viral entry as a strategy for broad-spectrum antivirals. F1000Research 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, E.; Vincent, M.J.; Wickham, L.; Hamelin, J.; Basak, A.; Nichol, S.T.; Chretien, M.; Seidah, N.G. Implication of proprotein convertases in the processing and spread of severe acute respiratory syndrome coronavirus. Biochem. Biophys. Res. Commun. 2005, 326, 554–563. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; Whittaker, G.R. Host cell entry of Middle East respiratory syndrome coronavirus after two-step, furin-mediated activation of the spike protein. Proc. Natl. Acad. Sci. USA 2014, 111, 15214–15219. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.W.; Chao, T.L.; Li, C.L.; Chiu, M.F.; Kao, H.C.; Wang, S.H.; Pang, Y.H.; Lin, C.H.; Tsai, Y.M.; Lee, W.H.; et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020, 33, 108254. [Google Scholar] [CrossRef]
- Wang, C.; Cheng, S.; Zhang, Y.; Ding, Y.; Chong, H.; Xing, H.; Jiang, S.; Li, X.; Ma, L. Long-Acting HIV-1 Fusion Inhibitory Peptides and their Mechanisms of Action. Viruses 2019, 11, 811. [Google Scholar] [CrossRef]
- Zhang, X.; Ding, X.; Zhu, Y.; Chong, H.; Cui, S.; He, J.; Wang, X.; He, Y. Structural and functional characterization of HIV-1 cell fusion inhibitor T20. AIDS 2019, 33, 1–11. [Google Scholar] [CrossRef]
- Ding, X.; Zhang, X.; Chong, H.; Zhu, Y.; Wei, H.; Wu, X.; He, J.; Wang, X.; He, Y. Enfuvirtide (T20)-Based Lipopeptide Is a Potent HIV-1 Cell Fusion Inhibitor: Implications for Viral Entry and Inhibition. J. Virol. 2017, 91, e00831-17. [Google Scholar] [CrossRef]
- Qiu, S.; Yi, H.; Hu, J.; Cao, Z.; Wu, Y.; Li, W. The binding mode of fusion inhibitor T20 onto HIV-1 gp41 and relevant T20-resistant mechanisms explored by computational study. Curr. HIV Res. 2012, 10, 182–194. [Google Scholar] [CrossRef]
- Marcink, T.C.; Yariv, E.; Rybkina, K.; Mas, V.; Bovier, F.T.; des Georges, A.; Greninger, A.L.; Alabi, C.A.; Porotto, M.; Ben-Tal, N.; et al. Hijacking the Fusion Complex of Human Parainfluenza Virus as an Antiviral Strategy. Mbio 2020, 11, e03203-19. [Google Scholar] [CrossRef]
- Outlaw, V.K.; Lemke, J.T.; Zhu, Y.; Gellman, S.H.; Porotto, M.; Moscona, A. Structure-Guided Improvement of a Dual HPIV3/RSV Fusion Inhibitor. J. Am. Chem. Soc. 2020, 142, 2140–2144. [Google Scholar] [CrossRef]
- Plattet, P.; Alves, L.; Herren, M.; Aguilar, H.C. Measles Virus Fusion Protein: Structure, Function and Inhibition. Viruses 2016, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Takada, A.; Watanabe, T.; Ito, H.; Kida, H.; Kawaoka, Y. Functional importance of the coiled-coil of the Ebola virus glycoprotein. J. Virol. 2000, 74, 10194–10201. [Google Scholar] [CrossRef] [PubMed]
- de Vries, R.D.; Schmitz, K.S.; Bovier, F.T.; Predella, C.; Khao, J.; Noack, D.; Haagmans, B.L.; Herfst, S.; Stearns, K.N.; Drew-Bear, J.; et al. Intranasal fusion inhibitory lipopeptide prevents direct-contact SARS-CoV-2 transmission in ferrets. Science 2021, 371, 1379–1382. [Google Scholar] [CrossRef]
- Outlaw, V.K.; Bovier, F.T.; Mears, M.C.; Cajimat, M.N.; Zhu, Y.; Lin, M.J.; Addetia, A.; Lieberman, N.A.P.; Peddu, V.; Xie, X.; et al. Inhibition of Coronavirus Entry In Vitro and Ex Vivo by a Lipid-Conjugated Peptide Derived from the SARS-CoV-2 Spike Glycoprotein HRC Domain. Mbio 2020, 11, e01935-20. [Google Scholar] [CrossRef] [PubMed]
- Lalezari, J.P.; Henry, K.; O’Hearn, M.; Montaner, J.S.; Piliero, P.J.; Trottier, B.; Walmsley, S.; Cohen, C.; Kuritzkes, D.R.; Eron, J.J., Jr.; et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N. Engl. J. Med. 2003, 348, 2175–2185. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, G.; Palmieri, F. Discovering genomic patterns in SARS-CoV-2 variants. Int. J. Intell. Syst. 2020, 35, 1680–1698. [Google Scholar] [CrossRef]
- Della Bella, E.; Koch, J.; Baerenfaller, K. Translation and emerging functions of non-coding RNAs in inflammation and immunity. Allergy 2022, 77, 2025–2037. [Google Scholar] [CrossRef]
- Tharakan, R.; Sawa, A. Minireview: Novel Micropeptide Discovery by Proteomics and Deep Sequencing Methods. Front. Genet. 2021, 12, 651485. [Google Scholar] [CrossRef]
- Koh, M.; Ahmad, I.; Ko, Y.; Zhang, Y.; Martinez, T.F.; Diedrich, J.K.; Chu, Q.; Moresco, J.J.; Erb, M.A.; Saghatelian, A.; et al. A short ORF-encoded transcriptional regulator. Proc. Natl. Acad. Sci. USA 2021, 118, e2021943118. [Google Scholar] [CrossRef]
- Slavoff, S.A.; Mitchell, A.J.; Schwaid, A.G.; Cabili, M.N.; Ma, J.; Levin, J.Z.; Karger, A.D.; Budnik, B.A.; Rinn, J.L.; Saghatelian, A. Peptidomic discovery of short open reading frame-encoded peptides in human cells. Nat. Chem. Biol. 2013, 9, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Cao, S.; Liu, X.; Huang, Y.; Yan, Y.; Zhou, C.; Shao, C.; Yang, R.; Zhu, W.; Du, Z.; Jia, C. Proteogenomic discovery of sORF-encoded peptides associated with bacterial virulence in Yersinia pestis. Commun. Biol. 2021, 4, 1248. [Google Scholar] [CrossRef] [PubMed]
- Diao, M.Q.; Li, C.; Xu, J.D.; Zhao, X.F.; Wang, J.X. RPS27, a sORF-Encoded Polypeptide, Functions Antivirally by Activating the NF-kappaB Pathway and Interacting With Viral Envelope Proteins in Shrimp. Front. Immunol. 2019, 10, 2763. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Mao, C.; Liu, S. Peptides encoded by noncoding genes: Challenges and perspectives. Signal Transduct. Target. Ther. 2019, 4, 57. [Google Scholar] [CrossRef] [PubMed]
- Caporale, A.; Doti, N.; Monti, A.; Sandomenico, A.; Ruvo, M. Automatic procedures for the synthesis of difficult peptides using oxyma as activating reagent: A comparative study on the use of bases and on different deprotection and agitation conditions. Peptides 2018, 102, 38–46. [Google Scholar] [CrossRef]
- Caporale, A.; Doti, N.; Sandomenico, A.; Ruvo, M. Evaluation of combined use of Oxyma and HATU in aggregating peptide sequences. J. Pept. Sci. 2017, 23, 272–281. [Google Scholar] [CrossRef]
- Zannella, C.; Chianese, A.; Palomba, L.; Marcocci, M.E.; Bellavita, R.; Merlino, F.; Grieco, P.; Folliero, V.; De Filippis, A.; Mangoni, M.; et al. Broad-Spectrum Antiviral Activity of the Amphibian Antimicrobial Peptide Temporin L and Its Analogs. Int. J. Mol. Sci. 2022, 23, 2060. [Google Scholar] [CrossRef]
- Sanna, G.; Madeddu, S.; Murgia, G.; Serreli, G.; Begala, M.; Caboni, P.; Incani, A.; Franci, G.; Galdiero, M.; Giliberti, G. Potent and Selective Activity against Human Immunodeficiency Virus 1 (HIV-1) of Thymelaea hirsuta Extracts. Viruses 2020, 12, 664. [Google Scholar] [CrossRef]
- Zhou, P.; Jin, B.; Li, H.; Huang, S.Y. HPEPDOCK: A web server for blind peptide-protein docking based on a hierarchical algorithm. Nucleic Acids Res. 2018, 46, W443–W450. [Google Scholar] [CrossRef]
- Ali, A.; Vijayan, R. Dynamics of the ACE2-SARS-CoV-2/SARS-CoV spike protein interface reveal unique mechanisms. Sci. Rep. 2020, 10, 14214. [Google Scholar] [CrossRef]
- Ali, N.; Khan, R.; AlAsmari, A.F.; Kumar, V. In silico investigations of heparin binding to SARS-CoV-2 variants with a focus at the RBD/ACE2 interface. Process Biochem. 2022, 115, 70–79. [Google Scholar] [CrossRef]
- Freidel, M.R.; Armen, R.S. Modeling the Structure-Activity Relationship of Arbidol Derivatives and Other SARS-CoV-2 Fusion Inhibitors Targeting the S2 Segment of the Spike Protein. J. Chem. Inf. Model. 2021, 61, 5906–5922. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Rawat, P.; Janakiraman, V.; Gromiha, M.M. Elucidating important structural features for the binding affinity of spike—SARS-CoV-2 neutralizing antibody complexes. Proteins 2022, 90, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Carino, A.; Moraca, F.; Fiorillo, B.; Marchiano, S.; Sepe, V.; Biagioli, M.; Finamore, C.; Bozza, S.; Francisci, D.; Distrutti, E.; et al. Hijacking SARS-CoV-2/ACE2 Receptor Interaction by Natural and Semi-synthetic Steroidal Agents Acting on Functional Pockets on the Receptor Binding Domain. Front. Chem. 2020, 8, 572885. [Google Scholar] [CrossRef]
- Mentlein, R. Cell-surface peptidases. Int. Rev. Cytol. 2004, 235, 165–213. [Google Scholar] [PubMed]
- Guan, H.; Chow, K.M.; Shah, R.; Rhodes, C.J.; Hersh, L.B. Degradation of islet amyloid polypeptide by neprilysin. Diabetologia 2012, 55, 2989–2998. [Google Scholar] [CrossRef] [PubMed]
- Sadremomtaz, A.; Al-Dahmani, Z.M.; Ruiz-Moreno, A.J.; Monti, A.; Wang, C.; Azad, T.; Bell, J.C.; Doti, N.; Velasco-Velazquez, M.A.; de Jong, D.; et al. Synthetic Peptides That Antagonize the Angiotensin-Converting Enzyme-2 (ACE-2) Interaction with SARS-CoV-2 Receptor Binding Spike Protein. J. Med. Chem. 2022, 65, 2836–2847. [Google Scholar] [CrossRef] [PubMed]
- Heitmann, J.S.; Bilich, T.; Tandler, C.; Nelde, A.; Maringer, Y.; Marconato, M.; Reusch, J.; Jager, S.; Denk, M.; Richter, M.; et al. A COVID-19 peptide vaccine for the induction of SARS-CoV-2 T cell immunity. Nature 2022, 601, 617–622. [Google Scholar] [CrossRef]
- Cabri, W.; Cantelmi, P.; Corbisiero, D.; Fantoni, T.; Ferrazzano, L.; Martelli, G.; Mattellone, A.; Tolomelli, A. Therapeutic Peptides Targeting PPI in Clinical Development: Overview, Mechanism of Action and Perspectives. Front. Mol. Biosci. 2021, 8, 697586. [Google Scholar] [CrossRef]
- Mahlapuu, M.; Hakansson, J.; Ringstad, L.; Bjorn, C. Antimicrobial Peptides: An Emerging Category of Therapeutic Agents. Front. Cell. Infect. Microbiol. 2016, 6, 194. [Google Scholar] [CrossRef]
- Fosgerau, K.; Hoffmann, T. Peptide therapeutics: Current status and future directions. Drug Discov. Today 2015, 20, 122–128. [Google Scholar] [CrossRef] [Green Version]
- Song, E.J.; Espano, E.; Shim, S.M.; Nam, J.H.; Kim, J.; Lee, K.; Park, S.K.; Lee, C.K.; Kim, J.K. Inhibitory effects of aprotinin on influenza A and B viruses in vitro and in vivo. Sci. Rep. 2021, 11, 9427. [Google Scholar] [CrossRef] [PubMed]
- Kaur, U.; Chakrabarti, S.S.; Ojha, B.; Pathak, B.K.; Singh, A.; Saso, L.; Chakrabarti, S. Targeting Host Cell Proteases to Prevent SARS-CoV-2 Invasion. Curr. Drug Targets 2021, 22, 192–201. [Google Scholar] [PubMed]
- Holly, M.K.; Diaz, K.; Smith, J.G. Defensins in Viral Infection and Pathogenesis. Annu. Rev. Virol. 2017, 4, 369–391. [Google Scholar] [CrossRef]
- Zhao, H.; Zhou, J.; Zhang, K.; Chu, H.; Liu, D.; Poon, V.K.; Chan, C.C.; Leung, H.C.; Fai, N.; Lin, Y.P.; et al. A novel peptide with potent and broad-spectrum antiviral activities against multiple respiratory viruses. Sci. Rep. 2016, 6, 22008. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Yang, D.; Li, W.; Wang, B.; Jiang, Z.; Li, M. Antiviral activity of recombinant mouse beta-defensin 3 against influenza A virus in vitro and in vivo. Antivir. Chem. Chemother. 2012, 22, 255–262. [Google Scholar] [CrossRef]
- Park, H.J.; Qin, H.; Cha, S.C.; Sharma, R.; Chung, Y.; Schluns, K.S.; Neelapu, S.S.; Overwijk, W.W.; Hwu, P.; Kwak, L.W. Induction of TLR4-dependent CD8+ T cell immunity by murine beta-defensin2 fusion protein vaccines. Vaccine 2011, 29, 3476–3482. [Google Scholar] [CrossRef] [PubMed]
- Gong, T.; Jiang, Y.; Wang, Y.; Yang, D.; Li, W.; Zhang, Q.; Feng, W.; Wang, B.; Jiang, Z.; Li, M. Recombinant mouse beta-defensin 2 inhibits infection by influenza A virus by blocking its entry. Arch. Virol. 2010, 155, 491–498. [Google Scholar] [CrossRef]
- Monteiro, A.; Yu, K.O.A.; Hicar, M.D. Peptide-based Fusion Inhibitors for Preventing the Six-helix Bundle Formation of Class I Fusion Proteins: HIV and Beyond. Curr. HIV Res. 2021, 19, 465–475. [Google Scholar] [CrossRef]
- Pu, J.; Zhou, J.T.; Liu, P.; Yu, F.; He, X.; Lu, L.; Jiang, S. Viral Entry Inhibitors Targeting Six-Helical Bundle Core against Highly Pathogenic Enveloped Viruses with Class I Fusion Proteins. Curr. Med. Chem. 2022, 29, 700–718. [Google Scholar] [CrossRef]
- Wang, C.; Xia, S.; Wang, X.; Li, Y.; Wang, H.; Xiang, R.; Jiang, Q.; Lan, Q.; Liang, R.; Li, Q.; et al. Supercoiling Structure-Based Design of a Trimeric Coiled-Coil Peptide with High Potency against HIV-1 and Human beta-Coronavirus Infection. J. Med. Chem. 2022, 65, 2809–2819. [Google Scholar] [CrossRef]
- Eggink, D.; Bontjer, I.; de Taeye, S.W.; Langedijk, J.P.M.; Berkhout, B.; Sanders, R.W. HIV-1 anchor inhibitors and membrane fusion inhibitors target distinct but overlapping steps in virus entry. J. Biol. Chem. 2019, 294, 5736–5746. [Google Scholar] [CrossRef] [PubMed]
- Pessi, A. Cholesterol-conjugated peptide antivirals: A path to a rapid response to emerging viral diseases. J. Pept. Sci. 2015, 21, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Porotto, M.; Yokoyama, C.C.; Palermo, L.M.; Mungall, B.; Aljofan, M.; Cortese, R.; Pessi, A.; Moscona, A. Viral entry inhibitors targeted to the membrane site of action. J. Virol. 2010, 84, 6760–6768. [Google Scholar] [CrossRef] [PubMed]
- Gustchina, E.; Louis, J.M.; Bewley, C.A.; Clore, G.M. Synergistic inhibition of HIV-1 envelope-mediated membrane fusion by inhibitors targeting the N and C-terminal heptad repeats of gp41. J. Mol. Biol. 2006, 364, 283–289. [Google Scholar] [CrossRef]
- Xu, Y.; Zhu, J.; Liu, Y.; Lou, Z.; Yuan, F.; Liu, Y.; Cole, D.K.; Ni, L.; Su, N.; Qin, L.; et al. Characterization of the heptad repeat regions, HR1 and HR2, and design of a fusion core structure model of the spike protein from severe acute respiratory syndrome (SARS) coronavirus. Biochemistry 2004, 43, 14064–14071. [Google Scholar] [CrossRef]
- Han, D.P.; Penn-Nicholson, A.; Cho, M.W. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology 2006, 350, 15–25. [Google Scholar] [CrossRef]
- Tesauro, D.; Accardo, A.; Diaferia, C.; Milano, V.; Guillon, J.; Ronga, L.; Rossi, F. Peptide-Based Drug-Delivery Systems in Biotechnological Applications: Recent Advances and Perspectives. Molecules 2019, 24, 351. [Google Scholar] [CrossRef]
- Bruno, B.J.; Miller, G.D.; Lim, C.S. Basics and recent advances in peptide and protein drug delivery. Ther. Deliv. 2013, 4, 1443–1467. [Google Scholar] [CrossRef]
- Moccia, G.; Carpinelli, L.; Savarese, G.; Borrelli, A.; Boccia, G.; Motta, O.; Capunzo, M.; De Caro, F. Perception of Health, Mistrust, Anxiety, and Indecision in a Group of Italians Vaccinated against COVID-19. Vaccines 2021, 9, 612. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SEQUENCE | SARS-CoV-2 | HCoV-229E | HCoV-OC43 | SARS-CoV-1 | MERS | HSV-1 | EA-71 | HIV-1 | YFV | WNV | DENV-2 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ACATTACAC | 25,372 | / | / | 25,247 | 25,227 | / | / | / | / | / | / |
| GTGTTTATT | 23,463 | / | 24,582 | / | / | / | / | / | / | / | / |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zannella, C.; Chianese, A.; Greco, G.; Santella, B.; Squillaci, G.; Monti, A.; Doti, N.; Sanna, G.; Manzin, A.; Morana, A.; et al. Design of Three Residues Peptides against SARS-CoV-2 Infection. Viruses 2022, 14, 2103. https://doi.org/10.3390/v14102103
Zannella C, Chianese A, Greco G, Santella B, Squillaci G, Monti A, Doti N, Sanna G, Manzin A, Morana A, et al. Design of Three Residues Peptides against SARS-CoV-2 Infection. Viruses. 2022; 14(10):2103. https://doi.org/10.3390/v14102103
Chicago/Turabian StyleZannella, Carla, Annalisa Chianese, Giuseppe Greco, Biagio Santella, Giuseppe Squillaci, Alessandra Monti, Nunzianna Doti, Giuseppina Sanna, Aldo Manzin, Alessandra Morana, and et al. 2022. "Design of Three Residues Peptides against SARS-CoV-2 Infection" Viruses 14, no. 10: 2103. https://doi.org/10.3390/v14102103
APA StyleZannella, C., Chianese, A., Greco, G., Santella, B., Squillaci, G., Monti, A., Doti, N., Sanna, G., Manzin, A., Morana, A., De Filippis, A., D’Angelo, G., Palmieri, F., Franci, G., & Galdiero, M. (2022). Design of Three Residues Peptides against SARS-CoV-2 Infection. Viruses, 14(10), 2103. https://doi.org/10.3390/v14102103