
Systems Bioinformatics Reveals Possible Relationship between COVID-19 and the Development of Neurological Diseases and Neuropsychiatric Disorders


Abstract
:1. Introduction
2. Methods
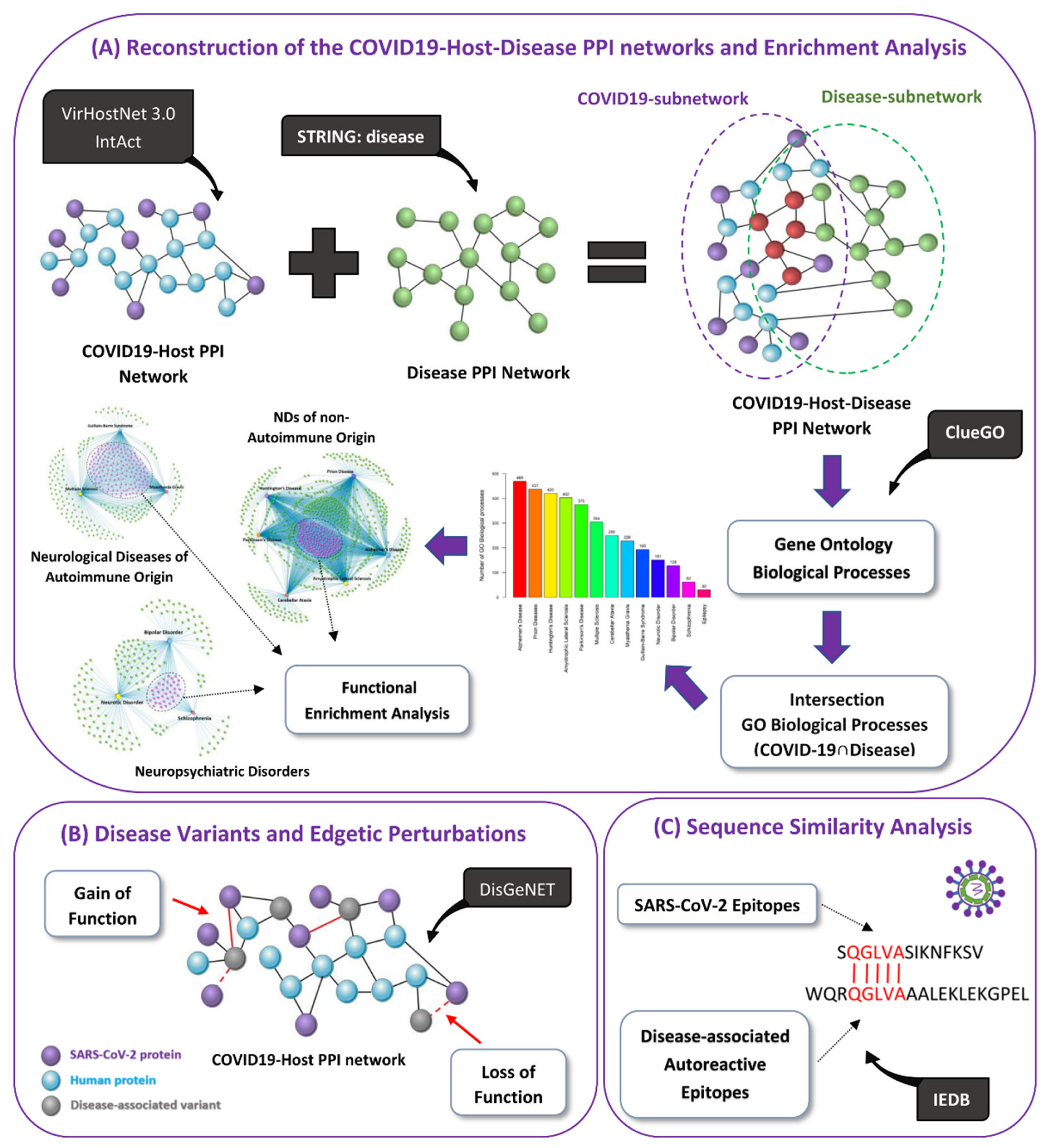
2.1. Network Reconstruction
2.1.1. Reconstruction of the COVID-19-Host PPI Network
2.1.2. Reconstruction of the Integrated COVID-19-Host-Disease PPI Networks
2.2. Enrichment Analysis
2.2.1. SARS-CoV-2 Human Protein Targets
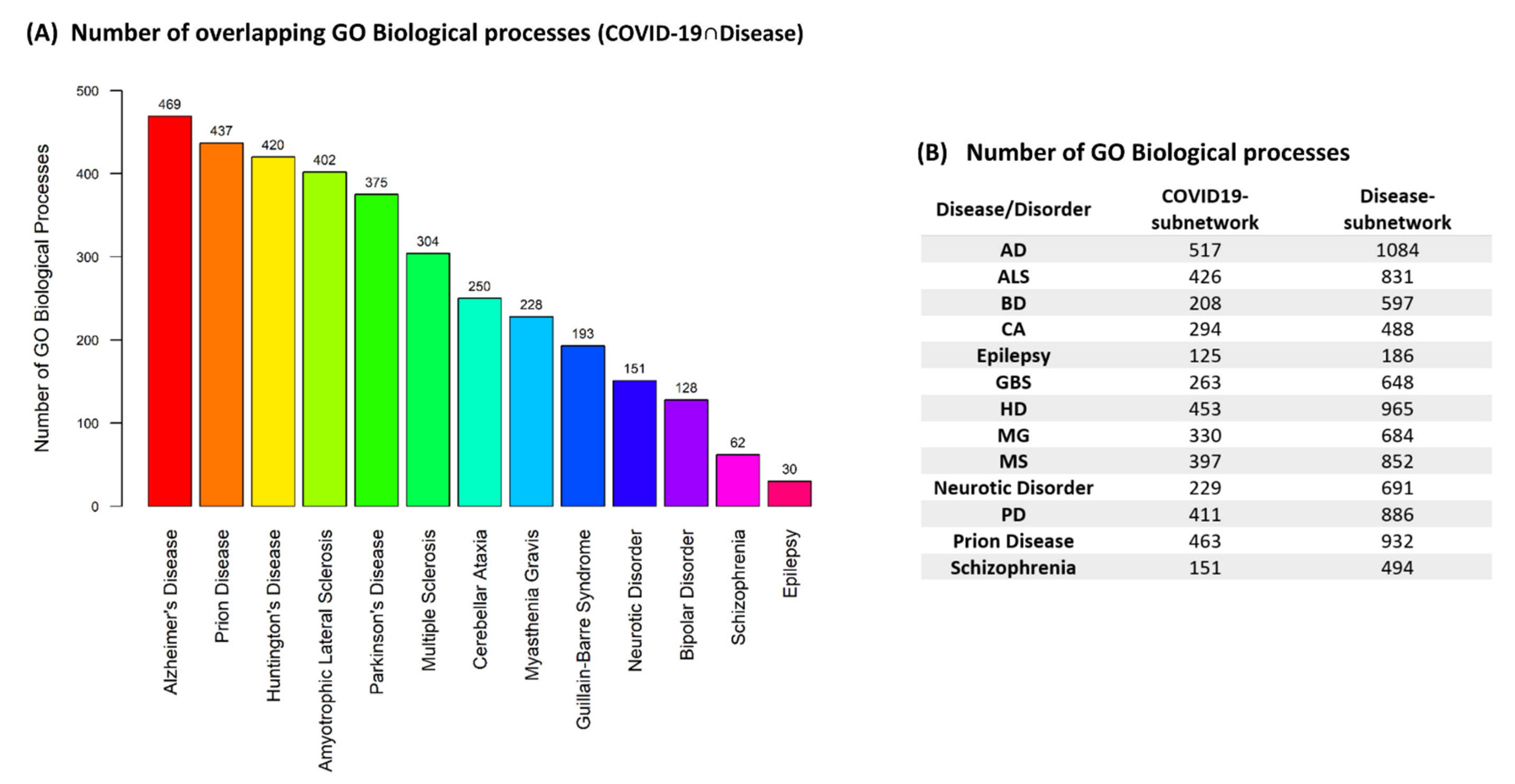
2.2.2. Overlapping GO Biological Processes (COVID-19 ∩ Disease)
2.3. Identification of Common GO Biological Processes Affected by COVID-19 in a Group of Diseases/Disorders
2.4. Identification of Disease-Associated Variants Targeted by SARS-CoV-2
2.5. Cross-Reactivity Potential via Molecular Mimicry of SARS-CoV-2 Epitopes
3. Results
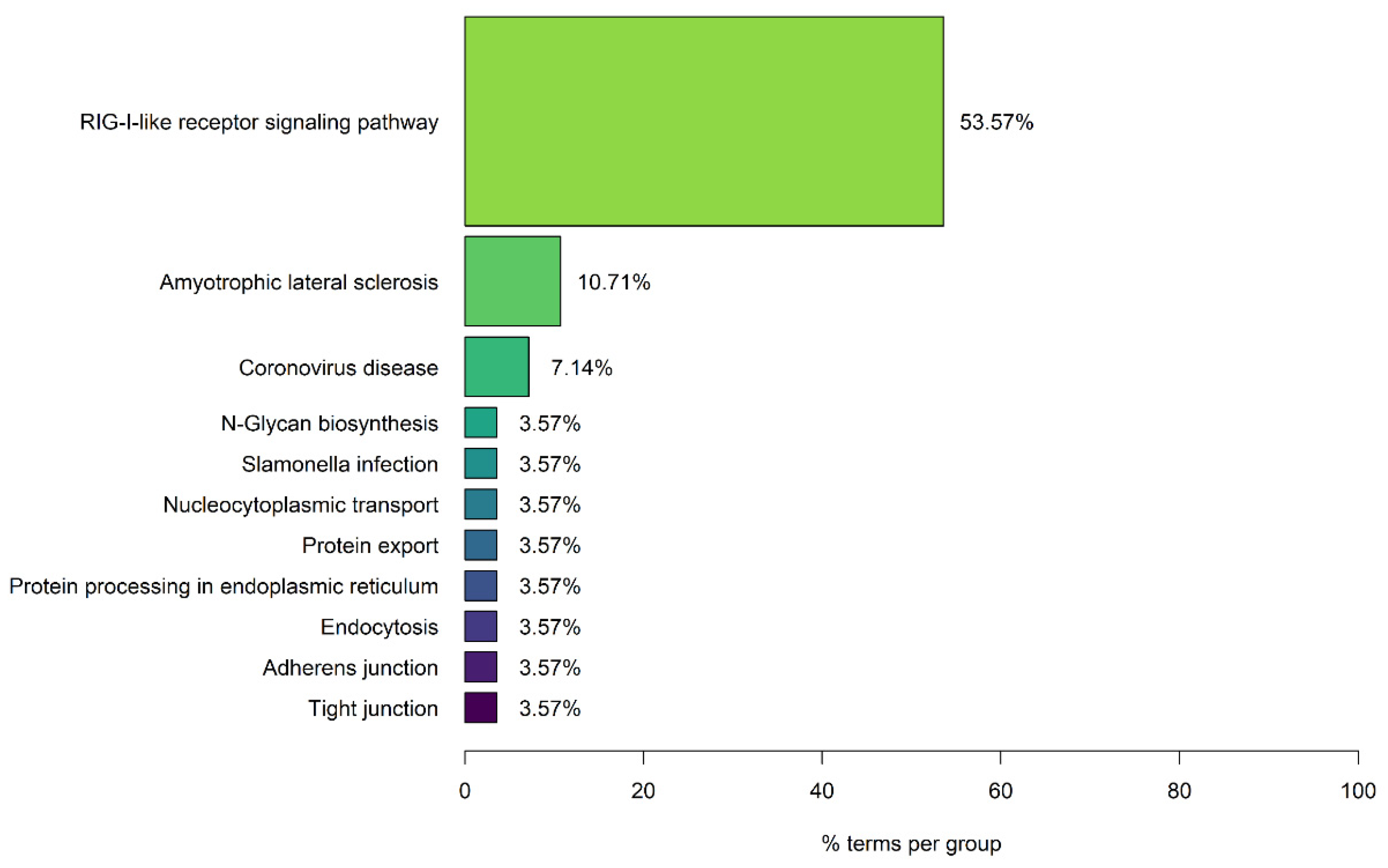
3.1. Enrichment Analysis Results of SARS-CoV-2 Human Protein Targets
3.2. Impact of SARS-CoV-2 Infection in Neurological Diseases and Neuropsychiatric Disorders
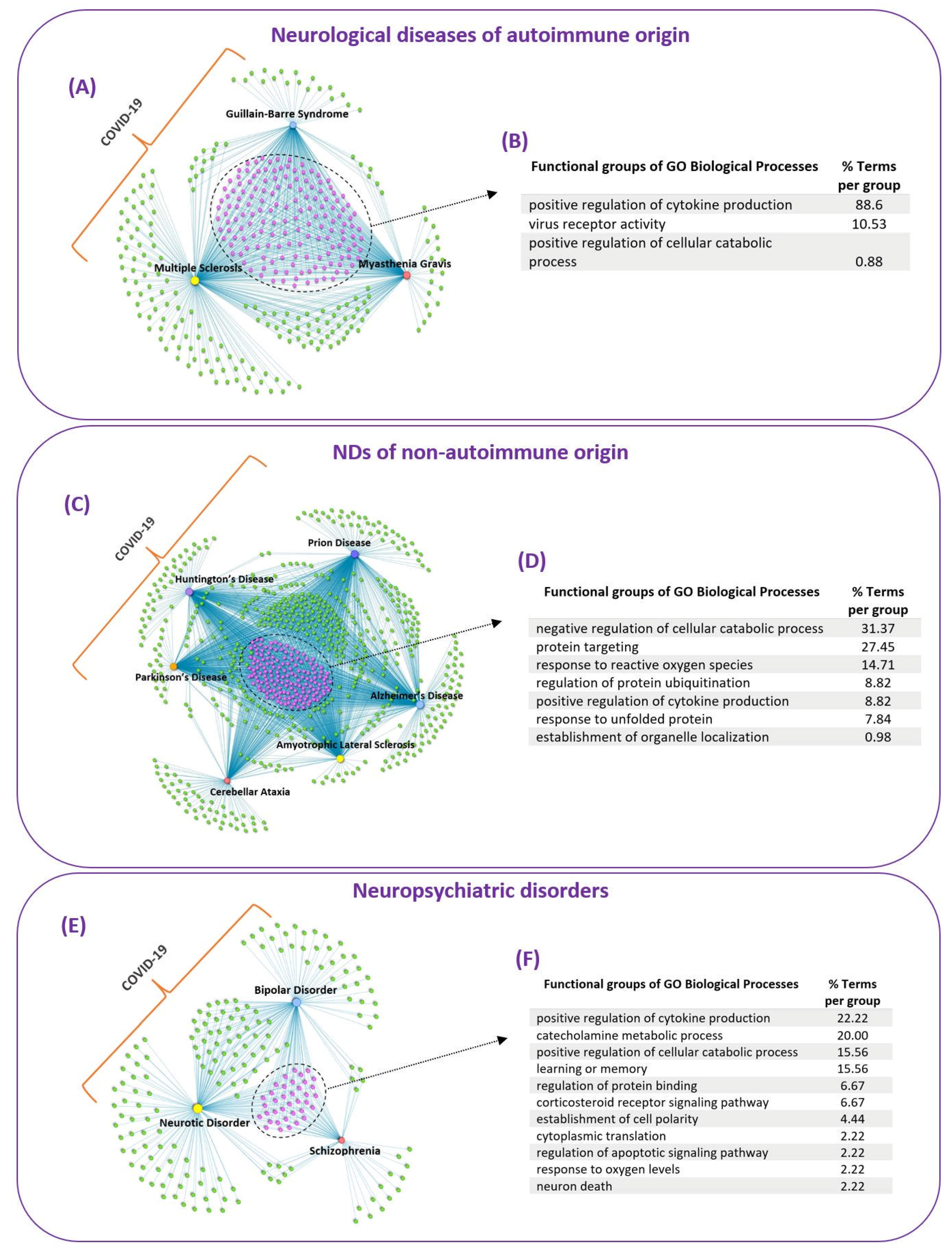
3.3. Common Mechanisms of Pathogenesis of SARS-CoV-2 Infection in Neurological Diseases and Neuropsychiatric Disorders
3.3.1. Neurological Diseases of Autoimmune Origin
3.3.2. NDs of Non-Autoimmune Origin
3.3.3. Neuropsychiatric Disorders
3.4. Disease-Associated Variant Genes/Proteins Interacting with SARS-CoV-2 Proteins
3.5. Cross-Reactivity Risk Potential of SARS-CoV-2 Epitopes Based on Similarity with Autoreactive Epitopes Found in Neurological Diseases
3.5.1. Matching 5-Mer Linear Motifs
3.5.2. Matching 6-Mer Linear Motifs
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chastain, E.M.L.; Miller, S.D. Molecular mimicry as an inducing trigger for CNS autoimmune demyelinating disease. Immunol. Rev. 2012, 245, 227–238. [Google Scholar] [CrossRef]
- Ercolini, A.M.; Miller, S.D. The role of infections in autoimmune disease. Clin. Exp. Immunol. 2009, 155, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Miranda-Saksena, M.; Saksena, N.K. Viruses and neurodegeneration. Virol. J. 2013, 10, 172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karim, S.; Mirza, Z.; Kamal, M.A.; Abuzenadah, A.M.; Azhar, E.I.; Al-Qahtani, M.H.; Damanhouri, G.A.; Ahmad, F.; Gan, S.H.; Sohrab, S.S. The Role of Viruses in Neurodegenerative and Neurobehavioral Diseases. CNS Neurol. Disord. Drug Targets 2014, 13, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Hogestyn, J.M.; Mock, D.J.; Mayer-Proschel, M. Contributions of neurotropic human herpesviruses herpes simplex virus 1 and human herpesvirus 6 to neurodegenerative disease pathology. Neural Regen. Res. 2018, 13, 211–221. [Google Scholar] [PubMed]
- Gonçalves de Andrade, E.; Šimončičová, E.; Carrier, M.; Vecchiarelli, H.A.; Robert, M.È.; Tremblay, M.È. Microglia Fighting for Neurological and Mental Health: On the Central Nervous System Frontline of COVID-19 Pandemic. Front. Cell. Neurosci. 2021, 15, 647378. [Google Scholar] [CrossRef] [PubMed]
- Ragab, D.; Salah Eldin, H.; Taeimah, M.; Khattab, R.; Salem, R. The COVID-19 Cytokine Storm; What We Know So Far. Front. Immunol. 2020, 11, 1446. [Google Scholar] [CrossRef]
- Tahaghoghi-Hajghorbani, S.; Zafari, P.; Masoumi, E.; Rajabinejad, M.; Jafari-Shakib, R.; Hasani, B.; Rafiei, A. The role of dysregulated immune responses in COVID-19 pathogenesis. Virus Res. 2020, 290, 198197. [Google Scholar] [CrossRef]
- Taquet, M.; Geddes, J.R.; Husain, M.; Luciano, S.; Harrison, P.J. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: A retrospective cohort study using electronic health records. Lancet Psychiatry 2021, 8, 416–427. [Google Scholar] [CrossRef]
- Mao, L.; Jin, H.; Wang, M.; Hu, Y.; Chen, S.; He, Q.; Chang, J.; Hong, C.; Zhou, Y.; Wang, D.; et al. Neurologic Manifestations of Hospitalized Patients with Coronavirus Disease 2019 in Wuhan, China. JAMA Neurol. 2020, 77, 683–690. [Google Scholar] [CrossRef]
- Serrano-Castro, P.J.; Estivill-Torrús, G.; Cabezudo-García, P.; Reyes-Bueno, J.A.; Ciano Petersen, N.; Aguilar-Castillo, M.J.; Suárez-Pérez, J.; Jiménez-Hernández, M.D.; Moya-Molina, M.Á.; Oliver-Martos, B.; et al. Impact of SARS-CoV-2 infection on neurodegenerative and neuropsychiatric diseases: A delayed pandemic? Neurología (English Edition) 2020, 35, 245–251. [Google Scholar] [CrossRef]
- Amruta, N.; Chastain, W.H.; Paz, M.; Solch, R.J.; Murray-Brown, I.C.; Befeler, J.B.; Gressett, T.E.; Longo, M.T.; Engler-Chiurazzi, E.B.; Bix, G. SARS-CoV-2 mediated neuroinflammation and the impact of COVID-19 in neurological disorders. Cytokine Growth Factor Rev. 2021, 58, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Guzman-Martinez, L.; Maccioni, R.B.; Andrade, V.; Navarrete, L.P.; Pastor, M.G.; Ramos-Escobar, N. Neuroinflammation as a common feature of neurodegenerative disorders. Front. Pharmacol. 2019, 10, 1008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Bedlack, R. COVID-19–accelerated disease progression in two patients with amyotrophic lateral sclerosis. Muscle Nerve 2021, 64, E13–E15. [Google Scholar] [CrossRef] [PubMed]
- Calistri, A.; Munegato, D.; Carli, I.; Parolin, C.; Palù, G. The ubiquitin-conjugating system: Multiple roles in viral replication and infection. Cells 2014, 3, 386–417. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, J.; Ploegh, H.L. Antigen Presentation and the Ubiquitin-Proteasome System in Host-Pathogen Interactions. Adv. Immunol. 2006, 92, 225–305. [Google Scholar] [PubMed]
- Zhao, Z.; Xia, J.; Tastan, O.; Singh, I.; Kshirsagar, M.; Carbonell, J.; Klein-Seetharaman, J. Virus interactions with human signal transduction pathways. Int. J. Comput. Biol. Drug Des. 2011, 4, 83–105. [Google Scholar] [CrossRef] [Green Version]
- Brito, A.F.; Pinney, J.W. Protein-protein interactions in virus-host systems. Front. Microbiol. 2017, 8, 1557. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.M.; Ernst, O.; Manes, N.P.; Oyler, B.L.; Fraser, I.D.C.; Goodlett, D.R.; Nita-Lazar, A. Multi-omics strategies uncover host-pathogen interactions. ACS Infect. Dis. 2019, 5, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Tekir, S.D.; Çakir, T.; Ülgen, K.Ö. Infection strategies of bacterial and viral pathogens through pathogen-human protein-protein interactions. Front. Microbiol. 2012, 3, 46. [Google Scholar]
- Bösl, K.; Ianevski, A.; Than, T.T.; Andersen, P.I.; Kuivanen, S.; Teppor, M.; Zusinaite, E.; Dumpis, U.; Vitkauskiene, A.; Cox, R.J.; et al. Common Nodes of Virus–Host Interaction Revealed Through an Integrated Network Analysis. Front. Immunol. 2019, 10, 2186. [Google Scholar] [CrossRef] [PubMed]
- Oulas, A.; Minadakis, G.; Zachariou, M.; Sokratous, K.; Bourdakou, M.M.; Spyrou, G.M. Systems Bioinformatics: Increasing precision of computational diagnostics and therapeutics through network-based approaches. Brief. Bioinform. 2017, 20, 806–824. [Google Scholar] [CrossRef] [PubMed]
- Onisiforou, A.; Spyrou, G.M. Identification of viral-mediated pathogenic mechanisms in neurodegenerative diseases using network-based approaches. Brief. Bioinform. 2021, 22, bbab141. [Google Scholar] [CrossRef] [PubMed]
- Onisiforou, A.; Spyrou, G.M. Immunomodulatory effects of microbiota-derived metabolites at the crossroad of neurodegenerative diseases and viral infection: Network-based bioinformatics insights. Front. Immunol. 2022, 13, 843128. [Google Scholar] [CrossRef]
- Geginat, J.; Paroni, M.; Pagani, M.; Galimberti, D.; De Francesco, R.; Scarpini, E.; Abrignani, S. The Enigmatic Role of Viruses in Multiple Sclerosis: Molecular Mimicry or Disturbed Immune Surveillance? Trends Immunol. 2017, 38, 498–512. [Google Scholar] [CrossRef]
- Riedhammer, C.; Weissert, R. Antigen presentation, autoantigens, and immune regulation in multiple sclerosis and other autoimmune diseases. Front. Immunol. 2015, 6, 322. [Google Scholar] [CrossRef] [Green Version]
- Waldner, H.; Collins, M.; Kuchroo, V.K. Activation of antigen-presenting cells by microbial products breaks self tolerance and induces autoimmune disease. J. Clin. Invest. 2004, 113, 990–997. [Google Scholar] [CrossRef] [Green Version]
- Oldstone, M.B.A. Molecular mimicry, microbial infection, and autoimmune disease: Evolution of the concept. In Current Topics in Microbiology and Immunology; Springer: Berlin/Heidelberg, Germany, 2005; Volume 296, pp. 1–17. [Google Scholar]
- Birnbaum, M.E.; Mendoza, J.L.; Sethi, D.K.; Dong, S.; Glanville, J.; Dobbins, J.; Özkan, E.; Davis, M.M.; Wucherpfennig, K.W.; Garcia, K.C. Deconstructing the peptide-MHC specificity of t cell recognition. Cell 2014, 157, 1073–1087. [Google Scholar] [CrossRef] [Green Version]
- van den Pol, A.N. Viral infections in the developing and mature brain. Trends Neurosci. 2006, 29, 398–406. [Google Scholar] [CrossRef]
- Patrick, K.L.; Bell, S.L.; Weindel, C.G.; Watson, R.O. Exploring the “multiple-hit hypothesis” of neurodegenerative disease: Bacterial infection comes up to bat. Front. Cell. Infect. Microbiol. 2019, 9, 138. [Google Scholar] [CrossRef] [Green Version]
- Deleidi, M.; Isacson, O. Viral and inflammatory triggers of neurodegenerative diseases. Sci. Transl. Med. 2012, 4, 121ps3. [Google Scholar] [CrossRef]
- Guirimand, T.; Delmotte, S.; Navratil, V. VirHostNet 2.0: Surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015, 43, D583–D587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerrien, S.; Aranda, B.; Breuza, L.; Bridge, A.; Broackes-Carter, F.; Chen, C.; Duesbury, M.; Dumousseau, M.; Feuermann, M.; Hinz, U.; et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012, 40, D841–D846. [Google Scholar] [CrossRef] [PubMed]
- Bartas, M.; Volná, A.; Beaudoin, C.A.; Poulsen, E.T.; Červeň, J.; Brázda, V.; Špunda, V.; Blundell, T.L.; Pečinka, P. Unheeded SARS-CoV-2 proteins? A deep look into negative-sense RNA. Brief. Bioinform. 2022, 23, bbac045. [Google Scholar] [CrossRef] [PubMed]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [Green Version]
- Stinissen, P.; Raus, J.; Zhang, J. Autoimmune pathogenesis of multiple sclerosis: Role of autoreactive T lymphocytes and new immunotherapeutic strategies. Crit. Rev. Immunol. 1997, 17, 33–75. [Google Scholar] [CrossRef]
- Wang, Z.; Yan, Y. Immunopathogenesis in myasthenia gravis and neuromyelitis optica. Front. Immunol. 2017. [CrossRef] [Green Version]
- Israeli, E.; Agmon-Levin, N.; Blank, M.; Chapman, J.; Shoenfeld, Y. Guillain-Barré syndrome-a classical autoimmune disease triggered by infection or vaccination. Clin. Rev. Allergy Immunol. 2012, 42, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Kwok, A.J.; Mentzer, A.; Knight, J.C. Host genetics and infectious disease: New tools, insights and translational opportunities. Nat. Rev. Genet. 2021, 22, 137–153. [Google Scholar] [CrossRef]
- Stojdl, D.F.; Lichty, B.D.; TenOever, B.R.; Paterson, J.M.; Power, A.T.; Knowles, S.; Marius, R.; Reynard, J.; Poliquin, L.; Atkins, H.; et al. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 2003, 4, 263–275. [Google Scholar] [CrossRef] [Green Version]
- Church, J.A. CCR5 Deficiency Increases Risk of Symptomatic West Nile Virus Infection. Pediatrics 2007, 120, S160. [Google Scholar] [CrossRef]
- Wicker, J.A.; Whiteman, M.C.; Beasley, D.W.C.; Davis, C.T.; Zhang, S.; Schneider, B.S.; Higgs, S.; Kinney, R.M.; Barrett, A.D.T. A single amino acid substitution in the central portion of the West Nile virus NS4B protein confers a highly attenuated phenotype in mice. Virology 2006, 349, 245–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daugherty, M.D.; Malik, H.S. Rules of engagement: Molecular insights from host-virus arms races. Annu. Rev. Genet. 2012, 46, 677–700. [Google Scholar] [CrossRef]
- Sahni, N.; Yi, S.; Zhong, Q.; Jailkhani, N.; Charloteaux, B.; Cusick, M.E.; Vidal, M. Edgotype: A fundamental link between genotype and phenotype. Curr. Opin. Genet. Dev. 2013, 23, 649–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulbahce, N.; Yan, H.; Dricot, A.; Padi, M.; Byrdsong, D.; Franchi, R.; Lee, D.S.; Rozenblatt-Rosen, O.; Mar, J.C.; Calderwood, M.A.; et al. Viral perturbations of host networks reflect disease etiology. PLoS Comput. Biol. 2012, 8, e1002531. [Google Scholar] [CrossRef]
- Piñero, J.; Queralt-Rosinach, N.; Bravo, À.; Deu-Pons, J.; Bauer-Mehren, A.; Baron, M.; Sanz, F.; Furlong, L.I. DisGeNET: A discovery platform for the dynamical exploration of human diseases and their genes. Database 2015, 2015, bav028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piñero, J.; Ramírez-Anguita, J.M.; Saüch-Pitarch, J.; Ronzano, F.; Centeno, E.; Sanz, F.; Furlong, L.I. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 2020, 48, D845–D855. [Google Scholar] [CrossRef] [Green Version]
- Vita, R.; Mahajan, S.; Overton, J.A.; Dhanda, S.K.; Martini, S.; Cantrell, J.R.; Wheeler, D.K.; Sette, A.; Peters, B. The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res. 2019, 47, D339–D343. [Google Scholar] [CrossRef] [Green Version]
- Pages, H.; Aboyoun, P.; Gentleman, R.; DebRoy, S. Biostrings: String objects representing biological sequences, and matching algorithms. R Package Version 2008, 2, 2008. [Google Scholar]
- Charif, D.; Lobry, J.R.; Necsulea, A.; Palmeira, L.; Perriere, G.; Penel, M.S. Package seqinr. R Package 2015, 218, bav028. [Google Scholar]
- Liu, Y.; Olagnier, D.; Lin, R. Host and viral modulation of RIG-I-mediated antiviral immunity. Front. Immunol. 2017, 7, 662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moudgil, K.D.; Choubey, D. Cytokines in autoimmunity: Role in induction, regulation, and treatment. J. Interf. Cytokine Res. 2011, 31, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Uzawa, A.; Kuwabara, S.; Suzuki, S.; Imai, T.; Murai, H.; Ozawa, Y.; Yasuda, M.; Nagane, Y.; Utsugisawa, K. Roles of cytokines and T cells in the pathogenesis of myasthenia gravis. Clin. Exp. Immunol. 2021, 203, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.K.; Blanco, M.R.; Bruce, E.A.; Honson, D.D.; Chen, L.M.; Chow, A.; Bhat, P.; Ollikainen, N.; Quinodoz, S.A.; Loney, C.; et al. SARS-CoV-2 Disrupts Splicing, Translation, and Protein Trafficking to Suppress Host Defenses. Cell 2020, 183, 1325–1339.e21. [Google Scholar] [CrossRef]
- Wenzhong, L.; Hualan, L. COVID-19: Captures iron and generates reactive oxygen species to damage the human immune system. Autoimmunity 2021, 54, 213–224. [Google Scholar] [CrossRef]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Aarli, J. Role of Cytokines in Neurological Disorders. Curr. Med. Chem. 2005, 10, 1931–1937. [Google Scholar] [CrossRef]
- Visser, D.; Golla, S.S.V.; Verfaillie, S.C.J.; Coomans, E.M.; Rikken, R.M.; Van De Giessen, E.M.; Den Hollander, M.E.; Verveen, A.; Yaqub, M.; Barkhof, F.; et al. Long COVID is associated with extensive in-vivo neuroinflammation on [18F]DPA-714 PET. medRxiv 2022. [Google Scholar] [CrossRef]
- Zheng, Q.; Huang, T.; Zhang, L.; Zhou, Y.; Luo, H.; Xu, H.; Wang, X. Dysregulation of ubiquitin-proteasome system in neurodegenerative diseases. Front. Aging Neurosci. 2016, 8, 303. [Google Scholar] [CrossRef] [Green Version]
- McKinnon, C.; Goold, R.; Andre, R.; Devoy, A.; Ortega, Z.; Moonga, J.; Linehan, J.M.; Brandner, S.; Lucas, J.J.; Collinge, J.; et al. Prion-mediated neurodegeneration is associated with early impairment of the ubiquitin–proteasome system. Acta Neuropathol. 2016, 131, 411–425. [Google Scholar] [CrossRef] [Green Version]
- Ortega, Z.; Lucas, J.J. Ubiquitin-proteasome system involvement in huntington’s disease. Front. Mol. Neurosci. 2014, 7, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, X.; Chen, Z.J. The role of ubiquitylation in immune defence and pathogen evasion. Nat. Rev. Immunol. 2012, 12, 35–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gustin, J.K.; Moses, A.V.; Früh, K.; Douglas, J.L. Viral takeover of the host ubiquitin system. Front. Microbiol. 2011, 2, 161. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zheng, H.; Zhu, J.; Dong, Q.; Wang, J.; Fan, H.; Chen, Y.; Zhang, X.; Han, X.; Li, Q.; et al. Ubiquitin-Modified Proteome of SARS-CoV-2-Infected Host Cells Reveals Insights into Virus-Host Interaction and Pathogenesis. J. Proteome Res. 2021, 20, 2224–2239. [Google Scholar] [CrossRef]
- Costela-Ruiz, V.J.; Illescas-Montes, R.; Puerta-Puerta, J.M.; Ruiz, C.; Melguizo-Rodríguez, L. SARS-CoV-2 infection: The role of cytokines in COVID-19 disease. Cytokine Growth Factor Rev. 2020, 54, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Najjar, S.; Pearlman, D.M.; Alper, K.; Najjar, A.; Devinsky, O. Neuroinflammation and psychiatric illness. J. Neuroinflammation 2013, 10, 816. [Google Scholar] [CrossRef] [Green Version]
- Radtke, F.A.; Chapman, G.; Hall, J.; Syed, Y.A. Modulating neuroinflammation to treat neuropsychiatric disorders. Biomed Res. Int. 2017, 2017, 5071786. [Google Scholar] [CrossRef] [Green Version]
- Frank, M.G.; Weber, M.D.; Watkins, L.R.; Maier, S.F. Stress-induced neuroinflammatory priming: A liability factor in the etiology of psychiatric disorders. Neurobiol. Stress 2016, 4, 62–70. [Google Scholar] [CrossRef] [Green Version]
- Ranjbar-Slamloo, Y.; Fazlali, Z. Dopamine and Noradrenaline in the Brain; Overlapping or Dissociate Functions? Front. Mol. Neurosci. 2020, 12, 334. [Google Scholar] [CrossRef] [Green Version]
- Søraas, A.; Bø, R.; Kalleberg, K.T.; Støer, N.C.; Ellingjord-Dale, M.; Landrø, N.I. Self-reported Memory Problems 8 Months after COVID-19 Infection. JAMA Netw. Open 2021, 4, 334. [Google Scholar] [CrossRef]
- Becker, J.H.; Lin, J.J.; Doernberg, M.; Stone, K.; Navis, A.; Festa, J.R.; Wisnivesky, J.P. Assessment of Cognitive Function in Patients after COVID-19 Infection. JAMA Netw. Open 2021, 4, e2130645. [Google Scholar] [CrossRef] [PubMed]
- Kapur, S.; John Mann, J. Role of the dopaminergic system in depression. Biol. Psychiatry 1992, 32, 1–17. [Google Scholar] [CrossRef]
- Jauhar, S.; Nour, M.M.; Veronese, M.; Rogdaki, M.; Bonoldi, I.; Azis, M.; Turkheimer, F.; McGuire, P.; Young, A.H.; Howes, O.D. A test of the transdiagnostic dopamine hypothesis of psychosis using positron emission tomographic imaging in bipolar affective disorder and schizophrenia. JAMA Psychiatry 2017, 74, 1206–1213. [Google Scholar] [CrossRef] [PubMed]
- Simanjuntak, Y.; Liang, J.J.; Lee, Y.L.; Lin, Y.L. Japanese encephalitis virus exploits dopamine D2 receptor-phospholipase C to target dopaminergic human neuronal cells. Front. Microbiol. 2017, 8, 651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaskill, P.J.; Yano, H.H.; Kalpana, G.V.; Javitch, J.A.; Berman, J.W. Dopamine receptor activation increases HIV entry into primary human macrophages. PLoS ONE 2014, 9, e108232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogata, A.; Tashiro, K.; Nukuzuma, S.; Nagashima, K.; Hall, W.W. A rat model of Parkinson’s disease induced by Japanese encephalitis virus. J. Neurovirol. 1997, 3, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamaue, N.; Ogata, A.; Terado, M.; Ohno, K.; Kikuchi, S.; Sasaki, H.; Tashiro, K.; Hirafuji, M.; Minami, M. Brain catecholamine alterations and pathological features with aging in parkinson disease model rat induced by japanese encephalitis virus. Neurochem. Res. 2006, 31, 1451–1455. [Google Scholar] [CrossRef] [PubMed]
- Misra, U.K.; Kalita, J.; Pandey, S.; Khanna, V.K.; Babu, G.N. Cerebrospinal fluid catecholamine levels in Japanese encephalitis patients with movement disorders. Neurochem. Res. 2005, 30, 1075–1078. [Google Scholar] [CrossRef] [PubMed]
- Yi, T.I.; Kim, B.K.; Ha, S.A.; Lim, J.Y.; Han, J.S. Neuropsychological and Psychiatric Impairment after West Nile Virus Encephalitis in Korean: A Case Report. Brain Neurorehabilit. 2014, 7, 131–135. [Google Scholar] [CrossRef]
- Sejvar, J.J. Clinical manifestations and outcomes of West Nile virus infection. Viruses 2014, 6, 606–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosanko, C.M.; Gilroy, J.; Wang, A.M.; Sanders, W.; Dulai, M.; Wilson, J.; Blum, K. West Nile virus encephalitis involving the substantia nigra: Neuroimaging and pathologic findings with literature review. Arch. Neurol. 2003, 60, 1448–1452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schafernak, K.T.; Bigio, E.H. West Nile virus encephalomyelitis with polio-like paralysis & nigral degeneration. Can. J. Neurol. Sci. 2006, 33, 407–410. [Google Scholar] [PubMed] [Green Version]
- Blázquez, A.B.; Martín-Acebes, M.A.; Saiz, J.C. Inhibition of West Nile virus multiplication in cell culture by anti-parkinsonian drugs. Front. Microbiol. 2016, 7, 296. [Google Scholar] [CrossRef] [PubMed]
- Stute, N.L.; Stickford, J.L.; Province, V.M.; Augenreich, M.A.; Ratchford, S.M.; Stickford, A.S.L. COVID-19 is getting on our nerves: Sympathetic neural activity and haemodynamics in young adults recovering from SARS-CoV-2. J. Physiol. 2021, 599, 4269–4285. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.T.; Holmes, S.E.; Pietrzak, R.H.; Esterlis, I. Neurobiology of Chronic Stress-Related Psychiatric Disorders: Evidence from Molecular Imaging Studies. Chronic Stress 2017, 1, 2470547017710916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhabhar, F.S. Enhancing versus suppressive effects of stress on immune function: Implications for immunoprotection and immunopathology. Neuroimmunomodulation 2009, 16, 300–317. [Google Scholar] [CrossRef] [Green Version]
- Elenkov, I.J. Effects of Catecholamines on the Immune Response. NeuroImmune Biol. 2007, 7, 189–206. [Google Scholar]
- Ince, L.M.; Weber, J.; Scheiermann, C. Control of leukocyte trafficking by stress-associated hormones. Front. Immunol. 2019, 10, 3143. [Google Scholar] [CrossRef] [Green Version]
- Flierl, M.A.; Rittirsch, D.; Huber-Lang, M.; Vidya Sarma, J.; Award, P. Catecholamines-Crafty weapons in the inflammatory arsenal of immune/inflammatory cells or opening Pandora’s box? Mol. Med. 2008, 14, 195–204. [Google Scholar] [CrossRef]
- Hodo, T.W.; de Aquino, M.T.P.; Shimamoto, A.; Shanker, A. Critical Neurotransmitters in the Neuroimmune Network. Front. Immunol. 2020, 11, 1869. [Google Scholar] [CrossRef]
- Miller, A.H.; Haroon, E.; Raison, C.L.; Felger, J.C. Cytokine targets in the brain: Impact on neurotransmitters and neurocircuits. Depress. Anxiety 2013, 30, 297–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sternberg, E.M. Neural regulation of innate immunity: A coordinated nonspecific host response to pathogens. Nat. Rev. Immunol. 2006, 6, 318–328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, M.; Ruggiero, A.; Squeglia, F.; Maga, G.; Berisio, R. A Structural View of SARS-CoV-2 RNA Replication Machinery: RNA Synthesis, Proofreading and Final Capping. Cells 2020, 9, 1267. [Google Scholar] [CrossRef] [PubMed]
- Vann, K.R.; Tencer, A.H.; Kutateladze, T.G. Inhibition of translation and immune responses by the virulence factor Nsp1 of SARS-CoV-2. Signal Transduct. Target. Ther. 2020, 5, 234. [Google Scholar] [CrossRef]
- Yuan, S.; Balaji, S.; Lomakin, I.B.; Xiong, Y. Coronavirus Nsp1: Immune Response Suppression and Protein Expression Inhibition. Front. Microbiol. 2021, 12, 752214. [Google Scholar] [CrossRef]
- Abdalla, A.E.; Xie, J.; Junaid, K.; Younas, S.; Elsaman, T.; Abosalif, K.O.A.; Alameen, A.A.M.; Mahjoob, M.O.; Elamir, M.Y.M.; Ejaz, H. Insight into the emerging role of sars-cov-2 nonstructural and accessory proteins in modulation of multiple mechanisms of host innate defense. Bosn. J. Basic Med. Sci. 2021, 21, 515–527. [Google Scholar] [CrossRef]
- Abdelhak, A.; Hottenrott, T.; Morenas-Rodríguez, E.; Suárez-Calvet, M.; Zettl, U.K.; Haass, C.; Meuth, S.G.; Rauer, S.; Otto, M.; Tumani, H.; et al. Glial Activation Markers in CSF and Serum From Patients With Primary Progressive Multiple Sclerosis: Potential of Serum GFAP as Disease Severity Marker? Front. Neurol. 2019, 10, 280. [Google Scholar] [CrossRef] [Green Version]
- Högel, H.; Rissanen, E.; Barro, C.; Matilainen, M.; Nylund, M.; Kuhle, J.; Airas, L. Serum glial fibrillary acidic protein correlates with multiple sclerosis disease severity. Mult. Scler. J. 2020, 26, 210–219. [Google Scholar] [CrossRef]
- Frontera, J.A.; Boutajangout, A.; Masurkar, A.V.; Betensky, R.A.; Ge, Y.; Vedvyas, A.; Debure, L.; Moreira, A.; Lewis, A.; Huang, J.; et al. Comparison of serum neurodegenerative biomarkers among hospitalized COVID-19 patients versus non-COVID subjects with normal cognition, mild cognitive impairment, or Alzheimer’s dementia. Alzheimer’s Dement. 2022, 18, 899–910. [Google Scholar] [CrossRef]
- Renaud, M.; Thibault, C.; Le Normand, F.; McDonald, E.G.; Gallix, B.; Debry, C.; Venkatasamy, A. Clinical Outcomes for Patients with Anosmia 1 Year after COVID-19 Diagnosis. JAMA Netw. Open 2021, 4, e2115352. [Google Scholar] [CrossRef]
- Lechien, J.R.; Chiesa-Estomba, C.M.; De Siati, D.R.; Horoi, M.; Le Bon, S.D.; Rodriguez, A.; Dequanter, D.; Blecic, S.; El Afia, F.; Distinguin, L.; et al. Olfactory and gustatory dysfunctions as a clinical presentation of mild-to-moderate forms of the coronavirus disease (COVID-19): A multicenter European study. Eur. Arch. Oto-Rhino-Laryngology 2020, 277, 2251–2261. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Anderson, A.J.; Stojanovski, D. Mitochondrial protein import dysfunction: Mitochondrial disease, neurodegenerative disease and cancer. FEBS Lett. 2021, 595, 1107–1131. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; El Maadidi, S.; Faletti, L.; Haun, F.; Labib, S.; Schejtman, A.; Maurer, U.; Borner, C. How do viruses control mitochondria-mediated apoptosis? Virus Res. 2015, 209, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Elesela, S.; Lukacs, N.W. Role of mitochondria in viral infections. Life 2021, 11, 232. [Google Scholar] [CrossRef]
- Ohta, A.; Nishiyama, Y. Mitochondria and viruses. Mitochondrion 2011, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ajaz, S.; McPhail, M.J.; Singh, K.K.; Mujib, S.; Trovato, F.M.; Napoli, S.; Agarwal, K. Mitochondrial metabolic manipulation by SARS-CoV-2 in peripheral blood mononuclear cells of patients with COVID-19. Am. J. Physiol. Cell Physiol. 2021, 320, C57–C65. [Google Scholar] [CrossRef]
- Schultz, J.L.; Nopoulos, P.C.; Gonzalez-Alegre, P. Human immunodeficiency virus infection in huntington’s disease is associated with an earlier age of symptom onset. J. Huntingtons. Dis. 2018, 7, 163–166. [Google Scholar] [CrossRef]
- Hampshire, A.; Trender, W.; Chamberlain, S.R.; Jolly, A.E.; Grant, J.E.; Patrick, F.; Mazibuko, N.; Williams, S.C.; Barnby, J.M.; Hellyer, P.; et al. Cognitive deficits in people who have recovered from COVID-19. eClinicalMedicine 2021, 39, 101044. [Google Scholar] [CrossRef]
- Matias-Guiu, J.A.; Delgado-Alonso, C.; Yus, M.; Polidura, C.; Gómez-Ruiz, N.; Valles-Salgado, M.; Ortega-Madueño, I.; Cabrera-Martín, M.N.; Matias-Guiu, J. “Brain Fog” by COVID-19 or Alzheimer’s Disease? A Case Report. Front. Psychol. 2021, 12, 5116. [Google Scholar] [CrossRef]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10. [Google Scholar] [CrossRef]
- Islam, M.T. Oxidative stress and mitochondrial dysfunction-linked neurodegenerative disorders. Neurol. Res. 2017, 39, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms Underlying Inflammation in Neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Q.; Liu, X.B.; Xu, Y.M.; Zhong, B.L. Understanding the psychiatric symptoms of COVID-19: A meta-analysis of studies assessing psychiatric symptoms in Chinese patients with and survivors of COVID-19 and SARS by using the Symptom Checklist-90-Revised. Transl. Psychiatry 2021, 11, 290. [Google Scholar] [CrossRef] [PubMed]
- Link, H. The cytokine storm in multiple sclerosis. Mult. Scler. 1998, 4, 12–15. [Google Scholar] [CrossRef]
- Jafarzadeh, A.; Chauhan, P.; Saha, B.; Jafarzadeh, S.; Nemati, M. Contribution of monocytes and macrophages to the local tissue inflammation and cytokine storm in COVID-19: Lessons from SARS and MERS, and potential therapeutic interventions. Life Sci. 2020, 257, 118102. [Google Scholar] [CrossRef]
- Wang, J.; Jiang, M.; Chen, X.; Montaner, L.J. Cytokine storm and leukocyte changes in mild versus severe SARS-CoV-2 infection: Review of 3939 COVID-19 patients in China and emerging pathogenesis and therapy concepts. J. Leukoc. Biol. 2020, 108, 17–41. [Google Scholar] [CrossRef]
- Roy, R.K.; Sharma, U.; Wasson, M.K.; Jain, A.; Hassan, M.I.; Prakash, H. Macrophage Activation Syndrome and COVID 19: Impact of MAPK Driven Immune-Epigenetic Programming by SARS-Cov-2. Front. Immunol. 2021, 12, 4162. [Google Scholar] [CrossRef]
- Toor, D.; Jain, A.; Kalhan, S.; Manocha, H.; Sharma, V.K.; Jain, P.; Tripathi, V.; Prakash, H. Tempering Macrophage Plasticity for Controlling SARS-CoV-2 Infection for Managing COVID-19 Disease. Front. Pharmacol. 2020, 11, 570698. [Google Scholar] [CrossRef]
- Giamarellos-Bourboulis, E.J.; Netea, M.G.; Rovina, N.; Akinosoglou, K.; Antoniadou, A.; Antonakos, N.; Damoraki, G.; Gkavogianni, T.; Adami, M.E.; Katsaounou, P.; et al. Complex Immune Dysregulation in COVID-19 Patients with Severe Respiratory Failure. Cell Host Microbe 2020, 27, 992–1000.e3. [Google Scholar] [CrossRef]
- Lin, C.I.; Yu, H.H.; Lee, J.H.; Wang, L.C.; Lin, Y.T.; Yang, Y.H.; Chiang, B.L. Clinical analysis of macrophage activation syndrome in pediatric patients with autoimmune diseases. Clin. Rheumatol. 2012, 31, 1223–1230. [Google Scholar] [CrossRef]
- Deane, S.; Selmi, C.; Teuber, S.S.; Gershwin, M.E. Macrophage activation syndrome in autoimmune disease. Int. Arch. Allergy Immunol. 2010, 153, 109–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shokri-Kojori, E.; Wang, G.J.; Wiers, C.E.; Demiral, S.B.; Guo, M.; Kim, S.W.; Lindgren, E.; Ramirez, V.; Zehra, A.; Freeman, C.; et al. β-Amyloid accumulation in the human brain after one night of sleep deprivation. Proc. Natl. Acad. Sci. USA 2018, 115, 4483–4488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibarra-Coronado, E.G.; Pantaleón-Martínez, A.M.; Velazquéz-Moctezuma, J.; Prospéro-García, O.; Méndez-Díaz, M.; Pérez-Tapia, M.; Pavón, L.; Morales-Montor, J. The Bidirectional Relationship between Sleep and Immunity against Infections. J. Immunol. Res. 2015, 2015, 678164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons-Weiler, J. Pathogenic priming likely contributes to serious and critical illness and mortality in COVID-19 via autoimmunity. J. Transl. Autoimmun. 2020, 3, 100051. [Google Scholar] [CrossRef]
- Gold, J.E.; Okyay, R.A.; Licht, W.E.; Hurley, D.J. Investigation of long covid prevalence and its relationship to epstein-barr virus reactivation. Pathogens 2021, 10, 763. [Google Scholar] [CrossRef]
- Franceschini, E.; Cozzi-Lepri, A.; Santoro, A.; Bacca, E.; Lancellotti, G.; Menozzi, M.; Gennari, W.; Meschiari, M.; Bedini, A.; Orlando, G.; et al. Herpes simplex virus re-activation in patients with SARS-CoV-2 pneumonia: A prospective, observational study. Microorganisms 2021, 9, 1896. [Google Scholar] [CrossRef]
- Bond, P. Ethnicity and the relationship between covid-19 and the herpes simplex viruses. Med. Hypotheses 2021, 146, 110447. [Google Scholar] [CrossRef]
- Zuo, T.; Zhang, F.; Lui, G.C.Y.; Yeoh, Y.K.; Li, A.Y.L.; Zhan, H.; Wan, Y.; Chung, A.C.K.; Cheung, C.P.; Chen, N.; et al. Alterations in Gut Microbiota of Patients With COVID-19 During Time of Hospitalization. Gastroenterology 2020, 159, 944–955.e8. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Zuo, T.; Lui, G.C.Y.; Zhang, F.; Liu, Q.; Li, A.Y.L.; Chung, A.C.K.; Cheung, C.P.; Tso, E.Y.K.; Fung, K.S.C.; et al. Gut microbiota composition reflects disease severity and dysfunctional immune responses in patients with COVID-19. Gut 2021, 70, 698–706. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef]
- Dinan, T.G.; Cryan, J.F. The Microbiome-Gut-Brain Axis in Health and Disease. Gastroenterol. Clin. North Am. 2017, 46, 77–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, W.R.; Shang, D.; Itzhaki, R.F. Neurotropic viruses and Alzheimer disease: Interaction of herpes simplex type I virus and apolipoprotein E in the etiology of the disease. Mol. Chem. Neuropathol. 1996, 28, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Langer-Gould, A.; Wu, J.; Lucas, R.; Smith, J.; Gonzales, E.; Amezcua, L.; Haraszti, S.; Chen, L.H.; Quach, H.; James, J.A.; et al. Epstein-Barr virus, cytomegalovirus, and multiple sclerosis susceptibility. Neurology 2017, 89, 1330–1337. [Google Scholar] [CrossRef] [Green Version]
- Abdoli, A.; Taghipour, A.; Pirestani, M.; Mofazzal Jahromi, M.A.; Roustazadeh, A.; Mir, H.; Ardakani, H.M.; Kenarkoohi, A.; Falahi, S.; Karimi, M. Infections, inflammation, and risk of neuropsychiatric disorders: The neglected role of “co-infection”. Heliyon 2020, 6, e05645. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
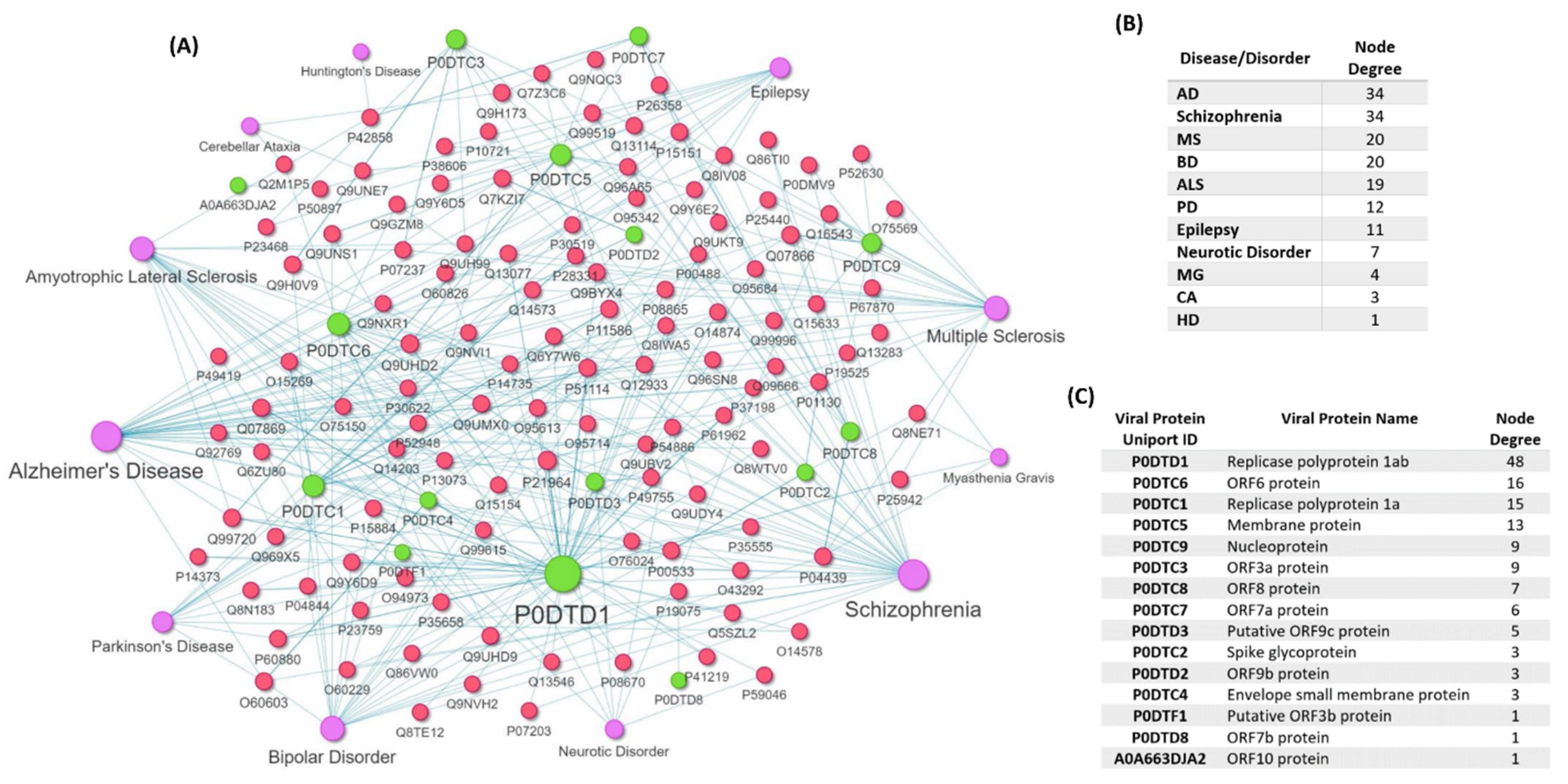{kind=link}
| Disease/Disorder | Number of Disease-Associated Variants from DisGeNET Database with Available Gene Name |
|---|---|
| ALS | 299 |
| CA | 87 |
| PD | 460 |
| AD | 737 |
| MG | 73 |
| HD | NA |
| MS | 536 |
| Prion Disease | 26 |
| GBS | 11 |
| BD | 402 |
| Epilepsy | 208 |
| Neurotic Disorder | 191 |
| Schizophrenia | 1179 |
| Viral Proteins Name | Viral Proteins Uniprot ID | Number of Linear Epitopes |
|---|---|---|
| ORF7b protein | P0DTD8 | 3 |
| ORF10 protein | A0A663DJA2 | 19 |
| ORF6 protein | P0DTC6 | 22 |
| Envelope small membrane protein | P0DTC4 | 24 |
| ORF7a protein | P0DTC7 | 49 |
| ORF8 protein | P0DTC8 | 87 |
| ORF3a protein | P0DTC3 | 171 |
| Membrane protein | P0DTC5 | 218 |
| Nucleoprotein | P0DTC9 | 512 |
| Spike glycoprotein | P0DTC2 | 1284 |
| Replicase polyprotein 1a | P0DTC1 | 1292 |
| Replicase polyprotein 1ab | P0DTD1 | 1315 |
| Total | 4996 |
| Neurological Disease | Number of Autoreactive Linear Epitopes | Number of Human Proteins Associated with Autoreactivity |
|---|---|---|
| MG | 284 | 10 |
| GBS | 11 | 5 |
| MS | 3512 | 1289 |
| AD | 62 | 6 |
| PD | 66 | 4 |
| ALS | 7 | 3 |
| Disease | Disease-Associated Autoreactive Epitopes | SARS-CoV-2 Epitopes | Consensus Sequence |
|---|---|---|---|
| AD | P05067-52 (APP) DAEFRHDSGYEVHHQKLVFFAQDVGSNKGAIIGLMVGGVV | P0DTD1-830 (Replicase polyprotein 1ab) ELKHFFFAQDGNAAI | K??FFAQD |
| AD | P05067-52 (APP) DAEFRHDSGYEVHHQKLVFFAQDVGSNKGAIIGLMVGGVV | P0DTD1-858 (Replicase polyprotein 1ab) FFFAQDGNAAISDYD | FFAQD |
| AD | P05067-52 (APP) DAEFRHDSGYEVHHQKLVFFAQDVGSNKGAIIGLMVGGVV | P0DTD1-1178 (Replicase polyprotein 1ab) SVELKHFFFAQDGNA | K??FFAQD |
| MG | P02708-64 (Acetylcholine receptor subunit alpha) LPTDSGEKMTLSISVLLSLTV | P0DTC1-21 (Replicase polyprotein 1a) KMVSLLSVLLSMQGA | KM????SVLLS? |
| MG | P07510-24 (Acetylcholine receptor subunit gamma) WQRQGLVAAALEKLEKGPEL | P0DTD1-685 (Replicase polyprotein 1ab) SQGLVASIKNFKSV | QGLVA? |
| PD | P10636-22 (MAPT) PKSPSSAKSRLQTAPV | P0DTD1-288 (Replicase polyprotein 1ab) EESSAKSASVY | SSAKS |
| PD | P10636-22 (MAPT) PKSPSSAKSRLQTAPV | P0DTD1-410 (Replicase polyprotein 1ab) SSAKSASVY | SSAKS |
| PD | P10636-22 (MAPT) PKSPSSAKSRLQTAPV | P0DTC1-64 (Replicase polyprotein 1a) CEESSAKSASVYYSQ | SSAKS |
| PD | P10636-22 (MAPT) PKSPSSAKSRLQTAPV | P0DTC1-330 (Replicase polyprotein 1a) EESSAKSASVYYSQL | SSAKS |
| PD | P10636-22 (MAPT) PKSPSSAKSRLQTAPV | P0DTC1-1178 (Replicase polyprotein 1a) VFDGKSKCEESSAKS | SSAKS |
| SARS-CoV-2 Viral Proteins Names | Uniprot ID of Human Protein Targets of MS-Associated Autoreactive Epitopes and Matching 5-Mer Motifs Found between SARS-CoV-2 Epitopes and MS Autoreactive Epitopes |
|---|---|
| P0DTC1 (Replicase polyprotein 1a) | J3QL64-[YLATA], P09543-[GKPVP], P02686-[YLATA], P10809-[IPKEE], P37837-[NYYKK], P60201-[ATLVS], P60201-[FFFLY], P04271-[NNELS], Q9H209-[VATVQ], P42701-[STVLS], P01042-[AVDAA], Q13571-[SKTPE], P11586-[QVNGL], P62861-[ARAGK], P62736-[EKSYE], P14921-[FITES], P04150-[EVVEN], Q06323-[DIILK], P05783-[LGSAL], P62316-[EEEEF], Q53YP1-[KDYLA], P08575-[KALRK], F1MPL6-[KDYLA], P05109-[LNSII], P29401-[YFDKA], Q05329-[QTTLK], A0A0D9SGF6-[RQGFV], P09972-[EASFN], A0A1W2PRT3-[DGEVI], P29401-[AEAEL], P08779-[AGALN], P08238-[SAFVE], Q13813-[KAGQK], O75508-[LALCA], P07196-[EEVLS], P09972-[VTALR], A0A0C4DGB6-[TFLKK], Q00653-[DFKLN], O43318-[DIAST], Q7L8L6-[TSSSK], Q15052-[ITGNT], P25440-[KKGAK], Q6ZV29-[KKPAS], Q8WXI7-[STVLS], P30153-[NVAKS], C9JXB8-[SFSGY], P18669-[GKAKK], O75486-[LLFLM], Q8MH48-[KDYLA], Q8NAP8-[VTDTP], Q9HCE3-[KLKAL], Q92688-[EEEEF], P0C7M7-[VSSPD], Q6V0I7-[GTGTI], P11487-[GIVAI], Q9Y6P5-[SEDAQ], A0A0C4DGB6-[LPSLA], P48039-[VLQVR], Q9BYE9-[LLVLV], P18858-[GKAKK], Q8NGD1-[DIQLL], P62854-[FDAYV], O95425-[LPTGV], Q5JNZ5-[FDAYV], Q9HC29-[LAKNV], Q96L11-[PILLL], Q9Y657-[RGMVL] |
| P0DTC2 (Spike glycoprotein) | P0DTU3-[FLLKY], O95674-[LNRAL], H7C2W9-[VTYVP], Q9NQ90-[GETGK], Q9BZR6-[LQELG], I3L0Y5-[GAGAA], O43318-[TNTSN], Q75MD7-[GAGAA], Q9ULK6-[GAGAA], P15880-[SYLTP], P05164-[DQLTP], P05164-[IVRFP], O75486-[EDLLF], Q8N446-[CCSCG], Q9NW61-[SFIED], Q6V0I7-[LTGTG], Q9NWB7-[KSNLK], Q9NTJ3-[VEAEV], Q9NTJ3-[KVEAE], Q13507-[VVLSF], Q15397-[AQEKN], Q5JNZ5-[VKLHY] |
| P0DTC3 (ORF3a protein) | Q6H3 × 3-[RATAT], Q9BYE9-[GVALL], P19875-[LLVAA] |
| P0DTC4 (Envelope small membrane protein) | P69905-[LVTLA] |
| P0DTC5 (Membrane protein) | P37108-[LLESE], P09972-[VLAAV], P09972-[LAAVY], Q5RI18-[KLLEQ], Q16695-[ARKSA] |
| P0DTC8 (ORF8 protein) | Q16695-[ARKSA], K7ERT8-[RKSAP], Q13630-[VGARK] |
| P0DTC9 (Nucleoprotein) | P10321-[ALLLL], Q13875-[SRGGS], A0A075B6H9-[PSASA], P05783-[SSSRS], P57073-[APSAS], P57073-[PSASA], P11161-[APSAS], P34903-[APSAS], Q8N729-[LALLL], Q8N729-[ALLLL], O95450-[ALLLL], O75486-[KDKKK], Q96GD3-[ALLLL], P82914-[YYRRA], Q6UX71-[VTQAF], P19875-[ALLLL], Q8IX21-[SSRSS] |
| P0DTD1 (Replicase polyprotein 1ab) | J3QL64-[YLATA], Q53Z42-[APRTL], P09543-[GKPVP], P02686-[YLATA], P37837-[NYYKK], A0A0A6YYK6-[SRQRL], Q8IUQ4-[LPTGT], Q6UWS5-[TVAGV], Q01082-[SSVEL], Q16663-[SVAAL], P14174-[VPRAS], P11586-[QVNGL], P14921-[FITES], P04150-[EVVEN], Q06323-[DIILK], P62316-[EEEEF], P47914-[TQAPT], P62937-[TVFFD], P06733-[ADLYK], Q05329-[LKYAI], Q05329-[QTTLK], P02489-[DDFVE], P09972-[EASFN], A0A1W2PRT3-[DGEVI], P08238-[LFENK], P29401-[AEAEL], P02511-[IRRPF], P10809-[KGVIT], P23297-[VAALT], Q86V81-[LDAYN], Q7L8L6-[TSSSK],Q6ZV29-[KKPAS], P30153-[RFNVA], P47914-[KPRSQ], C9JXB8-[SFSGY], P23528-[KEILV], P62851-[LLSKG], Q9UHN6-[YTFEK], Q96PV4-[KAVFI], P07355-[RDLYD], Q7Z4T9-[VYSFL], Q7Z4T9-[KYFVK],Q8NAP8-[VTDTP], Q92688-[EEEEF], P0C7M7-[VSSPD], P32246-[RARTV], P11487-[GIVAI], Q9Y6P5-[SEDAQ], Q8WXH0-[DTLKE], A0A0C4DGB6-[LPSLA], P62854-[FDAYV], O95425-[LPTGV], Q5JNZ5-[FDAYV], Q9HC29-[LAKNV], Q96L11-[PILLL] |
| Autoreactive Epitope Human Protein Target | Viral Epitope Protein Target | 6-Mer Motif |
|---|---|---|
| P10809 (60 kDa heat shock protein, mitochondrial) | P0DTD1 (Replicase polyprotein 1ab) | EIPKEE |
| P10809 (60 kDa heat shock protein, mitochondrial) | P0DTC1 (Replicase polyprotein 1a) | EIPKEE |
| P0DP02 (Immunoglobulin heavy variable 3-30-3) | P0DTD1 (Replicase polyprotein 1ab) | YYRARA |
| P0DP02 (Immunoglobulin heavy variable 3-30-3) | P0DTC1 (Replicase polyprotein 1a) | YYRARA |
| P62861 (40S ribosomal protein S30) | P0DTC1 (Replicase polyprotein 1a) | LARAGK |
| P62736 (Actin, aortic smooth muscle) | P0DTC1 (Replicase polyprotein 1a) | EKSYEL |
| P02489 (Alpha-crystallin A chain) | P0DTD1 (Replicase polyprotein 1ab) | DDFVEI |
| P09972 (Fructose-bisphosphate aldolase C) | P0DTC5 (Membrane protein (M)) | VLAAVY |
| Q16695 (Histone H3.1t) | P0DTC8 (ORF8 protein) | ARKSAP |
| P25440 (Bromodomain-containing protein 2) | P0DTD1 (Replicase polyprotein 1ab) | KKGAKL |
| P25440 (Bromodomain-containing protein 2) | P0DTC1 (Replicase polyprotein 1a) | KKGAKL |
| P57073 (Transcription factor SOX-8) | P0DTC9 (Nucleoprotein (N)) | APSASA |
| Q8N729 (Neuropeptide W) | P0DTC9 (Nucleoprotein (N)) | LALLLL |
| Q9NWB7 (Intraflagellar transport protein 57 homolog) | P0DTC2 (Spike glycoprotein (S)) | RKSNLK |
| Q9NTJ3 (Structural maintenance of chromosomes protein 4) | P0DTC2 (Spike glycoprotein (S)) | KVEAEV |
| P19875 (C-X-C motif chemokine 2) | P0DTC3 (ORF3a protein) | LLLVAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onisiforou, A.; Spyrou, G.M. Systems Bioinformatics Reveals Possible Relationship between COVID-19 and the Development of Neurological Diseases and Neuropsychiatric Disorders. Viruses 2022, 14, 2270. https://doi.org/10.3390/v14102270
Onisiforou A, Spyrou GM. Systems Bioinformatics Reveals Possible Relationship between COVID-19 and the Development of Neurological Diseases and Neuropsychiatric Disorders. Viruses. 2022; 14(10):2270. https://doi.org/10.3390/v14102270
Chicago/Turabian StyleOnisiforou, Anna, and George M. Spyrou. 2022. "Systems Bioinformatics Reveals Possible Relationship between COVID-19 and the Development of Neurological Diseases and Neuropsychiatric Disorders" Viruses 14, no. 10: 2270. https://doi.org/10.3390/v14102270
APA StyleOnisiforou, A., & Spyrou, G. M. (2022). Systems Bioinformatics Reveals Possible Relationship between COVID-19 and the Development of Neurological Diseases and Neuropsychiatric Disorders. Viruses, 14(10), 2270. https://doi.org/10.3390/v14102270