
Novel RNA Viruses Discovered in Weeds in Rice Fields


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. RNA Library Construction and Sequencing
2.3. RNA Virus Discovery
2.4. Virus Genome Annotation and Phylogenetic Analysis
2.5. Estimation of Viral Transcript Abundance and Virus Names
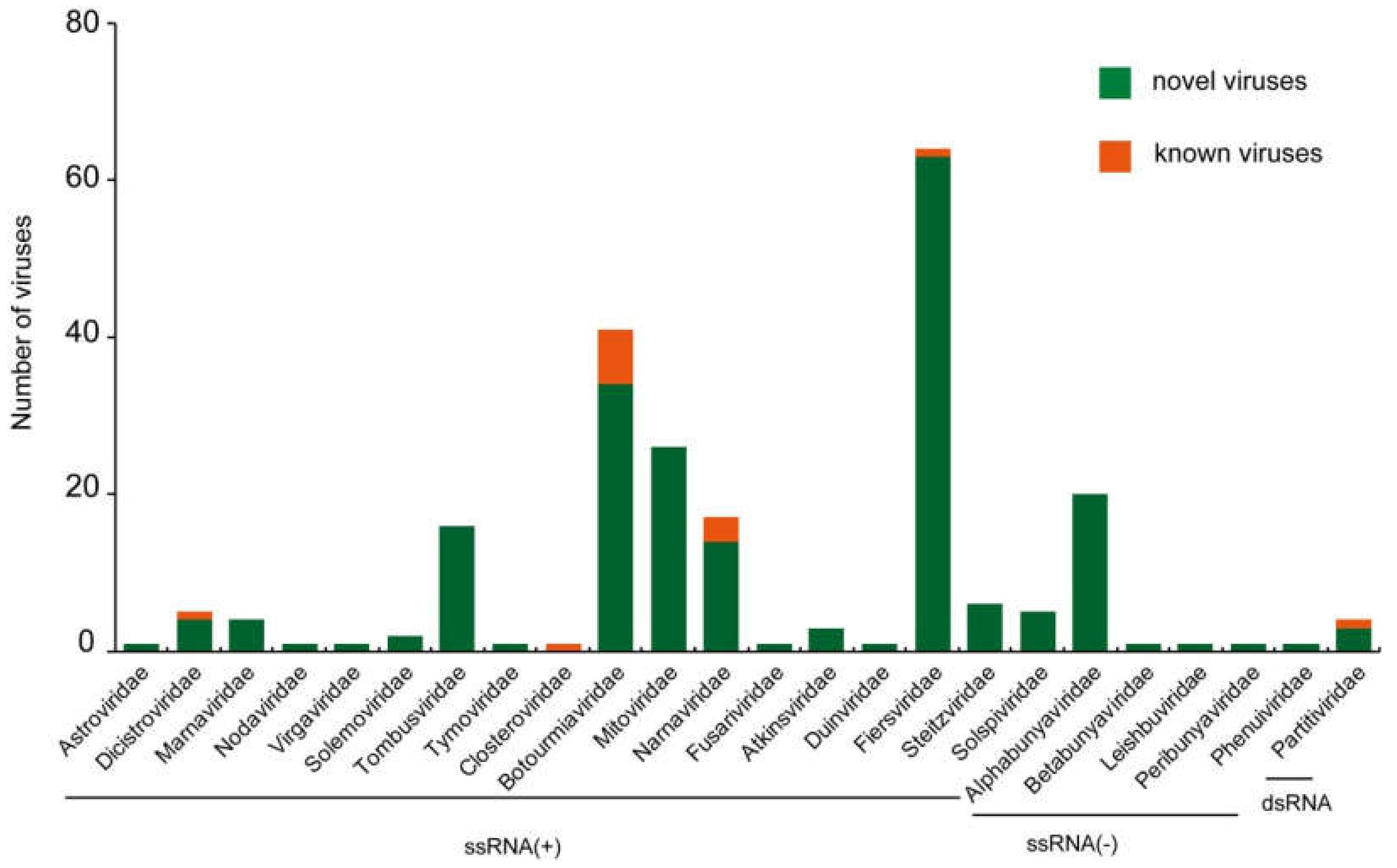
3. Results
3.1. Characterization of Postive-Sense RNA Viruses
3.2. Characterization of Negative-Sense RNA Viruses
3.3. Characterization of Double-Stranded RNA Viruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Power, A.G.; Mitchell, C.E. Pathogen Spillover in Disease Epidemics. Am. Nat. 2004, 164 (Suppl. 5), S79–S89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elena, S.F.; Fraile, A.; Garcia-Arenal, F. Evolution and emergence of plant viruses. Adv. Virus Res. 2014, 88, 161–191. [Google Scholar] [PubMed] [Green Version]
- Roossinck, M.J.; Garcia-Arenal, F. Ecosystem simplification, biodiversity loss and plant virus emergence. Curr. Opin. Virol. 2015, 10, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Marais, A.; Lefebvre, M.; Faure, C.; Candresse, T. Metagenomic analysis of virome cross-talk between cultivated Solanum lycopersicum and wild Solanum nigrum. Virology 2020, 540, 38–44. [Google Scholar] [CrossRef]
- Power, A.G.; Borer, E.T.; Hosseini, P.; Mitchell, C.E.; Seabloom, E.W. The community ecology of barley/cereal yellow dwarf viruses in Western US grasslands. Virus Res. 2011, 159, 95–100. [Google Scholar] [CrossRef]
- Bernardo, P.; Charles-Dominique, T.; Barakat, M.; Ortet, P.; Fernandez, E.; Filloux, D.; Hartnady, P.; Rebelo, T.A.; Cousins, S.R.; Mesleard, F.; et al. Geometagenomics illuminates the impact of agriculture on the distribution and prevalence of plant viruses at the ecosystem scale. ISME J. 2018, 12, 173–184. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.J. Plant Virus Metagenomics: Biodiversity and Ecology. Annu. Rev. Genet. 2012, 46, 359–369. [Google Scholar] [CrossRef]
- Hasiow-Jaroszewska, B.; Boezen, D.; Zwart, M.P. Metagenomic studies of viruses in weeds and wild plants: A powerful approach to characterise variable virus communities. Viruses 2021, 13, 1939. [Google Scholar] [CrossRef]
- Prendeville, H.R.; Ye, X.; Morris, T.J.; Pilson, D. Virus infections in wild plant populations are both frequent and often unapparent. Am. J. Bot. 2012, 99, 1033–1042. [Google Scholar] [CrossRef] [Green Version]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Yang, X.; Huang, J.; Liu, C.; Chen, B.; Zhang, T.; Zhou, G. Rice stripe mosaic virus, a novel cytorhabdovirus infecting rice via leafhopper transmission. Front. Microbiol. 2016, 7, 2140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Thompson, K.N.; Branck, T.; Yan, Y.; Nguyen, L.H.; Franzosa, E.A.; Huttenhower, C. Metatranscriptomics for the human microbiome and microbial community functional profiling. Annu. Rev. Biomed. Data Sci. 2021, 4, 279–311. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Chao, S.; Wang, H.; Yan, Q.; Chen, L.; Chen, G.; Wu, Y.; Meng, B.; Jin, L.; Zhu, X.; Feng, G. Metatranscriptomic sequencing suggests the presence of novel rna viruses in rice transmitted by brown planthopper. Viruses 2021, 13, 2464. [Google Scholar] [CrossRef]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.S.; Imrie, A.; Holmes, E.C. High-resolution metatranscriptomics reveals the ecological dynamics of mosquito-associated RNA viruses in Western Australia. J. Virol. 2017, 91, e00680-17. [Google Scholar] [CrossRef] [Green Version]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Nakashima, N.; Sasaki, J.; Toriyama, S. Determining the nucleotide sequence and capsid-coding region of himetobi P virus: A member of a novel group of RNA viruses that infect insects. Arch. Virol. 1999, 144, 2051–2058. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valles, S.M.; Chen, Y.; Firth, A.E.; Guerin, D.M.A.; Hashimoto, Y.; Herrero, S.; de Miranda, J.R.; Ryabov, E.; ICTV Report Consortium. ICTV virus taxonomy profile: Dicistroviridae. J. Gen. Virol. 2017, 98, 355–356. [Google Scholar] [CrossRef]
- Somera, M.; Fargette, D.; Hebrard, E.; Sarmiento, C.; ICTV Report Consortium. ICTV virus taxonomy profile: Solemoviridae 2021. J. Gen. Virol. 2021, 102, 001707. [Google Scholar] [CrossRef]
- Yassi, M.N.; Ritzenthaler, C.; Brugidou, C.; Fauquet, C.; Beachy, R.N. Nucleotide sequence and genome characterization of rice yellow mottle virus RNA. J. Gen. Virol. 1994, 75, 249–257. [Google Scholar] [CrossRef]
- Barrios Baron, M.P.; Agrofoglio, Y.C.; Delfosse, V.C.; Nahirnak, V.; Gonzalez de Urreta, M.; Almasia, N.I.; Vazquez Rovere, C.; Distefano, A.J. First complete genome sequence of potato leafroll virus from Argentina. Genome Announc. 2017, 5, e00628-17. [Google Scholar] [CrossRef] [Green Version]
- Moonan, F.; Molina, J.; Mirkov, T.E. Sugarcane yellow leaf virus: An emerging virus that has evolved by recombination between luteoviral and poleroviral ancestors. Virology 2000, 269, 156–171. [Google Scholar] [CrossRef]
- Tornos, T.; Cebrián, M.C.; Córdoba-Sellés, M.C.; Alfaro-Fernández, A.; Herrera-Vásquez, J.A.; Font, M.I.; Jorda, M.C. First report of pea enation mosaic virus infecting pea and broad bean in Spain. Plant Dis. 2008, 92, 1469. [Google Scholar] [CrossRef] [Green Version]
- Silva, J.M.F.; Al Rwahnih, M.; Blawid, R.; Nagata, T.; Fajardo, T.V.M. Discovery and molecular characterization of a novel enamovirus, Grapevine enamovirus-1. Virus Genes 2017, 53, 667–671. [Google Scholar] [CrossRef]
- Bejerman, N.; Giolitti, F.; Trucco, V.; De Breuil, S.; Dietzgen, R.G.; Lenardon, S. Complete genome sequence of a new enamovirus from Argentina infecting alfalfa plants showing dwarfism symptoms. Arch. Virol. 2016, 161, 2029–2032. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, Y.; Liu, W.; Cao, M.; Massart, S.; Wang, X. Identification, characterization and full-length sequence analysis of a novel polerovirus associated with wheat leaf yellowing disease. Front. Microbiol. 2017, 8, 1689. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.; Meliopoulos, V.A.; Karlsson, E.A.; Hargest, V.; Johnson, C.; Schultz-Cherry, S. Astrovirus biology and pathogenesis. Annu. Rev. Virol. 2017, 4, 327–348. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Hargest, V.; Cortez, V.; Meliopoulos, V.A.; Schultz-Cherry, S. Astrovirus pathogenesis. Viruses 2017, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Lukashov, V.V.; Goudsmit, J. Evolutionary relationships among Astroviridae. J. Gen. Virol. 2002, 83, 1397–1405. [Google Scholar] [CrossRef]
- Vlok, M.; Lang, A.S.; Suttle, C.A. Application of a sequence-based taxonomic classification method to uncultured and unclassified marine single-stranded RNA viruses in the order Picornavirales. Virus Evol. 2019, 5, vez056. [Google Scholar] [CrossRef]
- Chen, L.; Anane, R.F.; Wang, Z.; Chen, Z.; Gao, L.; Wen, G.; Zhao, M. Characterization of a novel Tombusviridae species isolated from Paris polyphylla var. yunnanensis. Arch. Virol. 2021, 166, 3199–3205. [Google Scholar] [CrossRef]
- Boonham, N.; Henry, C.M.; Wood, K.R. The nucleotide sequence and proposed genome organization of oat chlorotic stunt virus, a new soil-borne virus of cereals. J. Gen. Virol. 1995, 76, 2025–2034. [Google Scholar] [CrossRef]
- Sahul Hameed, A.S.; Ninawe, A.S.; Nakai, T.; Chi, S.C.; Johnson, K.L.; ICTV Report Consortium. ICTV virus taxonomy profile: Nodaviridae. J. Gen. Virol. 2019, 100, 3–4. [Google Scholar] [CrossRef]
- Martelli, G.P.; Sabanadzovic, S.; Abou-Ghanem Sabanadzovic, N.; Edwards, M.C.; Dreher, T. The family Tymoviridae. Arch. Virol. 2002, 147, 1837–1846. [Google Scholar] [CrossRef]
- Yang, L.; Zhang, S.; Mei, S.; Liu, Q.; Zhou, Y.; Li, R.; Cao, M. Complete genome sequence of a novel citrus virus with characteristics of members of the family Tymoviridae. Arch. Virol. 2021, 166, 2055–2058. [Google Scholar] [CrossRef] [PubMed]
- Maccheroni, W.; Alegria, M.C.; Greggio, C.C.; Piazza, J.P.; Kamla, R.F.; Zacharias, P.R.; Bar-Joseph, M.; Kitajima, E.W.; Assumpcao, L.C.; Camarotte, G.; et al. Identification and genomic characterization of a new virus (Tymoviridae family) associated with citrus sudden death disease. J. Virol. 2005, 79, 3028–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, M.J.; Adkins, S.; Bragard, C.; Gilmer, D.; Li, D.; MacFarlane, S.A.; Wong, S.M.; Melcher, U.; Ratti, C.; Ryu, K.H.; et al. ICTV virus taxonomy profile: Virgaviridae. J. Gen. Virol. 2017, 98, 1999–2000. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, M.; Bar-Joseph, M.; Candresse, T.; Maree, H.J.; Martelli, G.P.; Melzer, M.J.; Menzel, W.; Minafra, A.; Sabanadzovic, S.; ICTV Report Consortium. ICTV virus taxonomy profile: Closteroviridae. J. Gen. Virol. 2020, 101, 364–365. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Shi, X.; Shi, J.; Zhang, Z.; Fang, Y.; Zhang, Z.; Pan, Q.; Zheng, L.; Gao, Y.; Zhang, D.; et al. Tomato chlorosis virus infection facilitates bemisia tabaci med reproduction by elevating vitellogenin expression. Insects 2021, 12, 101–112. [Google Scholar] [CrossRef]
- Ayllon, M.A.; Turina, M.; Xie, J.; Nerva, L.; Marzano, S.L.; Donaire, L.; Jiang, D.; ICTV Report Consortium. ICTV virus taxonomy profile: Botourmiaviridae. J. Gen. Virol. 2020, 101, 454–455. [Google Scholar] [CrossRef]
- Oses-Ruiz, M.; Cruz-Mireles, N.; Martin-Urdiroz, M.; Soanes, D.M.; Eseola, A.B.; Tang, B.; Derbyshire, P.; Nielsen, M.; Cheema, J.; Were, V.; et al. Appressorium-mediated plant infection by Magnaporthe oryzae is regulated by a Pmk1-dependent hierarchical transcriptional network. Nat. Microbiol. 2021, 6, 1383–1397. [Google Scholar] [CrossRef]
- Chen, X.; Hai, D.; Li, J.; Tan, J.; Huang, S.; Zhang, H.; Chen, H.; Zhang, M. Complete genome sequence of a novel mitovirus associated with Lagenaria siceraria. Arch. Virol. 2021, 166, 3427–3431. [Google Scholar] [CrossRef]
- Shahi, S.; Eusebio-Cope, A.; Kondo, H.; Hillman, B.I.; Suzuki, N. Investigation of host range of and host defense against a mitochondrially replicating mitovirus. J. Virol. 2019, 93, e01503-18. [Google Scholar] [CrossRef] [Green Version]
- Hillman, B.I.; Cai, G. The family Narnaviridae: Simplest of RNA viruses. Adv. Virus Res. 2013, 86, 149–176. [Google Scholar]
- Zhang, R.; Liu, S.; Chiba, S.; Kondo, H.; Kanematsu, S.; Suzuki, N. A novel single-stranded RNA virus isolated from a phytopathogenic filamentous fungus, Rosellinia necatrix, with similarity to hypo-like viruses. Front. Microbiol. 2014, 5, 360. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Cai, L.; Liu, M.; Wang, X.; Yang, J.; An, H.; Deng, Q.; Zhang, S.; Fang, S. A novel previously undescribed fusarivirus from the phytopathogenic fungus Setosphaeria turcica. Arch. Virol. 2021, 166, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Callanan, J.; Stockdale, S.R.; Adriaenssens, E.M.; Kuhn, J.H.; Rumnieks, J.; Pallen, M.J.; Shkoporov, A.N.; Draper, L.A.; Ross, R.P.; Hill, C. Leviviricetes: Expanding and restructuring the taxonomy of bacteria-infecting single-stranded RNA viruses. Microb. Genom. 2021, 7, 000686. [Google Scholar] [CrossRef] [PubMed]
- Herath, V.; Romay, G.; Urrutia, C.D.; Verchot, J. Family level phylogenies reveal relationships of plant viruses within the order bunyavirales. Viruses 2020, 12, 1010. [Google Scholar] [CrossRef] [PubMed]
- Vainio, E.J.; Chiba, S.; Ghabrial, S.A.; Maiss, E.; Roossinck, M.; Sabanadzovic, S.; Suzuki, N.; Xie, J.; Nibert, M.; ICTV Report Consortium. ICTV virus taxonomy profile: Partitiviridae. J. Gen. Virol. 2018, 99, 17–18. [Google Scholar] [CrossRef]
- Gadhave, K.R.; Gautam, S.; Rasmussen, D.A.; Srinivasan, R. Aphid transmission of potyvirus: The largest plant-infecting RNA virus genus. Viruses 2020, 12, 773. [Google Scholar] [CrossRef]
- Gupta, N.; Reddy, K.; Bhattacharyya, D.; Chakraborty, S. Plant responses to geminivirus infection: Guardians of the plant immunity. Virol. J. 2021, 18, 143. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, S.; Mei, S.; Zhou, Y.; Wang, J.; Han, G.Z.; Chen, L.; Zhou, C.; Cao, M. Viromics unveils extraordinary genetic diversity of the family Closteroviridae in wild citrus. PLoS Pathog. 2021, 17, e1009751. [Google Scholar] [CrossRef]
- Wang, Z.; Neupane, A.; Feng, J.; Pedersen, C.; Lee Marzano, S.Y. Direct metatranscriptomic survey of the sunflower microbiome and virome. Viruses 2021, 13, 1867. [Google Scholar] [CrossRef]
- Lee, H.J.; Jeong, R.D. Metatranscriptomic analysis of plant viruses in imported pear and kiwifruit pollen. Plant Pathol. J. 2022, 38, 220–228. [Google Scholar] [CrossRef]
- Choi, H.; Jo, Y.; Cho, W.K.; Yu, J.; Tran, P.T.; Salaipeth, L.; Kwak, H.R.; Choi, H.S.; Kim, K.H. Identification of viruses and viroids infecting tomato and pepper plants in vietnam by metatranscriptomics. Int. J. Mol. Sci. 2020, 21, 7565. [Google Scholar] [CrossRef] [PubMed]
- Dion, M.B.; Oechslin, F.; Moineau, S. Phage diversity, genomics and phylogeny. Nat. Rev. Microbiol. 2020, 18, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Jover, L.F.; Cortez, M.H.; Weitz, J.S. Mechanisms of multi-strain coexistence in host-phage systems with nested infection networks. J. Theor. Biol. 2013, 332, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Bondy-Denomy, J.; Qian, J.; Westra, E.R.; Buckling, A.; Guttman, D.S.; Davidson, A.R.; Maxwell, K.L. Prophages mediate defense against phage infection through diverse mechanisms. ISME J. 2016, 10, 2854–2866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dedrick, R.M.; Jacobs-Sera, D.; Bustamante, C.A.; Garlena, R.A.; Mavrich, T.N.; Pope, W.H.; Reyes, J.C.; Russell, D.A.; Adair, T.; Alvey, R.; et al. Prophage-mediated defence against viral attack and viral counter-defence. Nat. Microbiol. 2017, 2, 16251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, S.; Afzal, M.; Iqbal, S.; Khan, Q.M. Plant-bacteria partnerships for the remediation of hydrocarbon contaminated soils. Chemosphere 2013, 90, 1317–1332. [Google Scholar] [CrossRef] [PubMed]
- Smith-Moore, C.M.; Grunden, A.M. Bacteria and archaea as the sources of traits for enhanced plant phenotypes. Biotechnol. Adv. 2018, 36, 1900–1916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Y.X.; Zhang, N.; Hu, B.; Jin, T.; Xu, H.; Qin, Y.; Yan, P.; Zhang, X.; Guo, X.; et al. NRT1.1B is associated with root microbiota composition and nitrogen use in field-grown rice. Nat. Biotechnol. 2019, 37, 676–684. [Google Scholar]
- Kovalskaya, N.; Foster-Frey, J.; Donovan, D.M.; Bauchan, G.; Hammond, R.W. Antimicrobial activity of bacteriophage endolysin produced in nicotiana benthamiana plants. J. Microbiol. Biotechnol. 2016, 26, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Rahimi-Midani, A.; Choi, T.J. Transport of phage in melon plants and inhibition of progression of bacterial fruit blotch. Viruses 2020, 12, 477. [Google Scholar] [CrossRef] [Green Version]
- Roossinck, M.J. Viruses in the phytobiome. Curr. Opin. Virol. 2019, 37, 72–76. [Google Scholar] [CrossRef]
- Hillman, B.I.; Annisa, A.; Suzuki, N. Viruses of plant-interacting fungi. Adv. Virus Res. 2018, 100, 99–116. [Google Scholar] [PubMed]
- Yan, L.; Zhu, J.; Zhao, X.; Shi, J.; Jiang, C.; Shao, D. Beneficial effects of endophytic fungi colonization on plants. Appl. Microbiol. Biotechnol. 2019, 103, 3327–3340. [Google Scholar] [CrossRef] [PubMed]
- Lo Presti, L.; Lanver, D.; Schweizer, G.; Tanaka, S.; Liang, L.; Tollot, M.; Zuccaro, A.; Reissmann, S.; Kahmann, R. Fungal effectors and plantsusceptibility. Annu. Rev. Plant Biol. 2015, 66, 513–545. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chao, S.; Wang, H.; Zhang, S.; Chen, G.; Mao, C.; Hu, Y.; Yu, F.; Wang, S.; Lv, L.; Chen, L.; et al. Novel RNA Viruses Discovered in Weeds in Rice Fields. Viruses 2022, 14, 2489. https://doi.org/10.3390/v14112489
Chao S, Wang H, Zhang S, Chen G, Mao C, Hu Y, Yu F, Wang S, Lv L, Chen L, et al. Novel RNA Viruses Discovered in Weeds in Rice Fields. Viruses. 2022; 14(11):2489. https://doi.org/10.3390/v14112489
Chicago/Turabian StyleChao, Shufen, Haoran Wang, Shu Zhang, Guoqing Chen, Chonghui Mao, Yang Hu, Fengquan Yu, Shuo Wang, Liang Lv, Long Chen, and et al. 2022. "Novel RNA Viruses Discovered in Weeds in Rice Fields" Viruses 14, no. 11: 2489. https://doi.org/10.3390/v14112489
APA StyleChao, S., Wang, H., Zhang, S., Chen, G., Mao, C., Hu, Y., Yu, F., Wang, S., Lv, L., Chen, L., & Feng, G. (2022). Novel RNA Viruses Discovered in Weeds in Rice Fields. Viruses, 14(11), 2489. https://doi.org/10.3390/v14112489