
Rhinovirus Infection and Virus-Induced Asthma

 , ,
, , 
Abstract
:1. Introduction
2. Pathophysiology of Asthma
3. Major Processes of Airway Tissue Remodelling in Virus-Induced Asthma
4. RV Virology
5. Obstacles to Understanding Cytokines in Virus-Induced Asthma
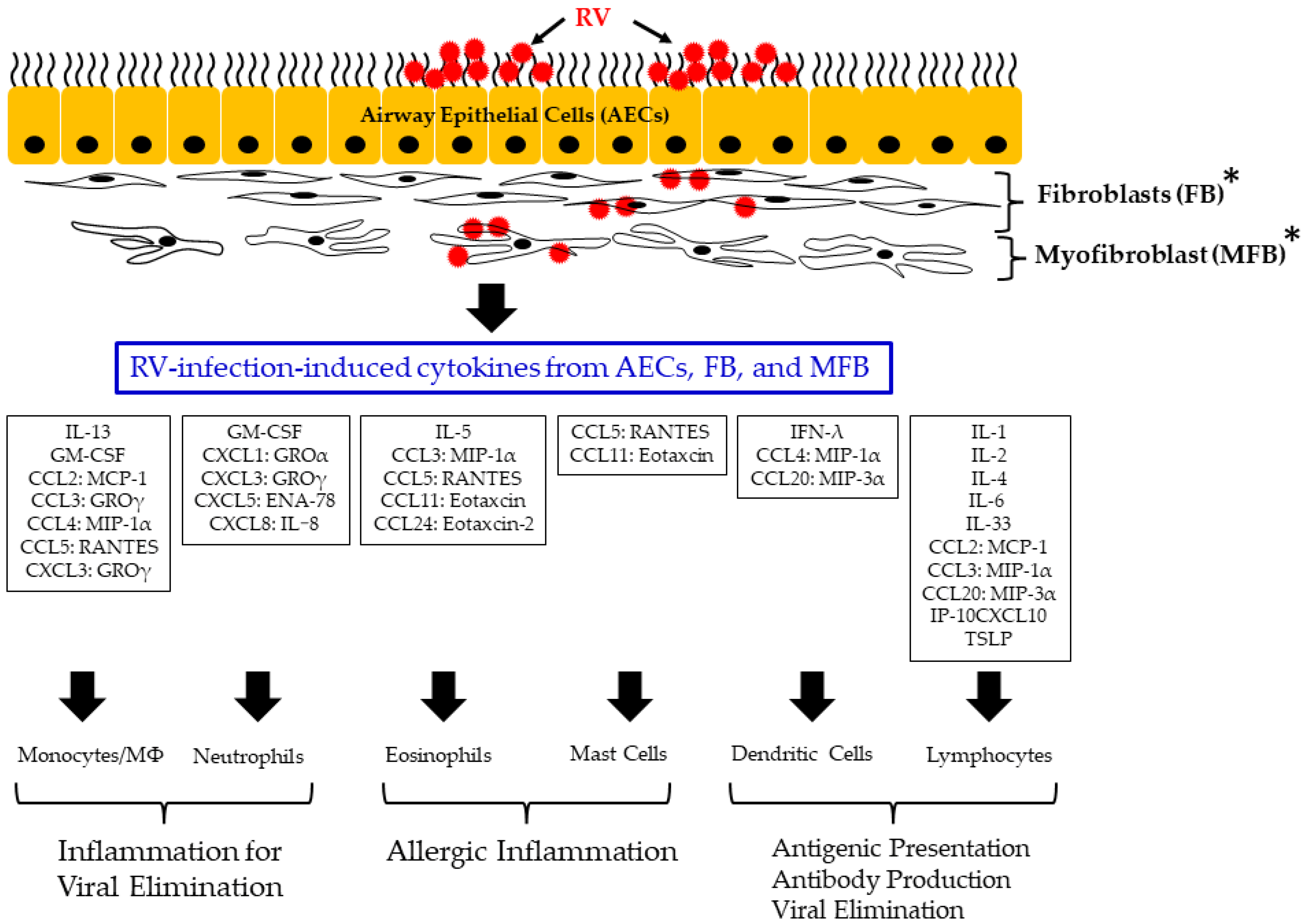
6. Relationships between RV-Infection-Induced Cytokine Responses in Airway Epithelial Cells, Fibroblasts, and Myofibroblasts
7. Effector Functions of Leukocytes
8. Differences between Asthmatics and Non-Asthmatics in Viral Infection
9. Perspective
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Kusel, M.M.H.; de Klerk, N.H.; Kebadze, T.; Vohma, V.; Holt, P.G.; Johnston, S.L.; Sly, P.D. Early-life respiratory viral infections, atopic sensitization, and risk of subsequent development of persistent asthma. J. Allergy Clin. Immunol. 2007, 119, 1105–1110. [Google Scholar] [CrossRef]
- Johnston, N.W.; Sears, M.R. Asthma exacerbations 1: Epidemiology. Thorax 2006, 61, 722–728. [Google Scholar] [CrossRef] [Green Version]
- Kurai, D.; Saraya, T.; Ishii, H.; Takizawa, H. Virus-induced exacerbations in asthma and COPD. Front. Microbiol. 2013, 4, 293. [Google Scholar] [CrossRef] [Green Version]
- Khetsuriani, N.; Lu, X.; Teague, W.G.; Kazerouni, N.; Anderson, L.J.; Erdman, D.D. Novel human rhinoviruses and exacerbation of asthma in children. Emerg. Infect. Dis. 2008, 14, 1793. [Google Scholar] [CrossRef]
- Humberto, E.T.B.; Samuel, A. Histology for Pathologists; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2019. [Google Scholar]
- Toews, G.B. Cytokines and the lung. Eur. Respir. J. 2001, 34, 3s–17s. [Google Scholar] [CrossRef] [Green Version]
- Gern, J.E. Mechanisms of virus-induced asthma. J. Pediatr. 2003, 142, S9–S13. [Google Scholar] [CrossRef] [PubMed]
- Bousquet, J.; Jeffery, P.K.; Busse, W.W.; Johnson, M.; Vignola, A.M. Asthma: From bronchoconstriction to airways inflammation and remodeling. Am. J. Respir. Crit. Care Med. 2000, 161, 1720–1745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Muhsen, S.; Johnson, J.R.; Hamid, Q. Remodeling in asthma. J. Allergy Clin. Immunol. 2011, 128, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Czarnowicki, T.; Esaki, H.; Gonzalez, J.; Malajian, D.; Shemer, A.; Noda, S.; Talasila, S.; Berry, A.; Gray, J.; Becker, L. Early pediatric atopic dermatitis shows only a cutaneous lymphocyte antigen (CLA)+ TH2/TH1 cell imbalance, whereas adults acquire CLA+ TH22/TC22 cell subsets. J. Allergy Clin. Immunol. 2015, 136, 941–951. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Yoshizumi, M.; Ishii, H.; Oishi, K.; Ryo, A. Cytokine production and signaling pathways in respiratory virus infection. Front. Microbiol. 2013, 4, 276. [Google Scholar] [CrossRef]
- James, E.; Gern, A.C. Fields Virology, 6th Edition. Clin. Infect. Dis. 2013, 59, 531–549. [Google Scholar] [CrossRef]
- Jacobs, S.E.; Lamson, D.M.; St George, K.; Walsh, T.J. Human rhinoviruses. Clin. Microbiol. Rev. 2013, 26, 135–162. [Google Scholar] [CrossRef] [Green Version]
- McIntyre, C.L.; Knowles, N.J.; Simmonds, P. Proposals for the classification of human rhinovirus species A, B and C into genotypically assigned types. J. Gen. Virol. 2013, 94, 1791. [Google Scholar] [CrossRef] [PubMed]
- Alshrari, A.S.; Hudu, S.A.; Asdaq, S.M.; Ali, A.M.; Kin, C.V.; Omar, A.R.; Pei, C.P.; Sekawi, Z. Bioinformatics analysis of rhinovirus capsid proteins VP1–4 sequences for cross-serotype vaccine development. J. Infect. Public Health 2021, 14, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- McWilliam Leitch, E.C.; Cabrerizo, M.; Cardosa, J.; Harvala, H.; Ivanova, O.E.; Kroes, A.C.; Lukashev, A.; Muir, P.; Odoom, J.; Roivainen, M.; et al. Evolutionary dynamics and temporal/geographical correlates of recombination in the human enterovirus echovirus types 9, 11, and 30. J. Virol. 2010, 84, 9292–9300. [Google Scholar] [CrossRef] [Green Version]
- Basnet, S.; Palmenberg, A.C.; Gern, J.E. Rhinoviruses and their receptors. Chest 2019, 155, 1018–1025. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.-H.; Pang, L.-L.; Yang, J.; Jin, Y. Comparison of immune response to human rhinovirus C and respiratory syncytial virus in highly differentiated human airway epithelial cells. Virol. J. 2022, 19, 81. [Google Scholar] [CrossRef]
- Romagnani, S. Human TH1 and TH2 subsets: Doubt no more. Immunol. Today 1991, 12, 256–257. [Google Scholar] [CrossRef] [PubMed]
- Zuniga, E.I.; Macal, M.; Lewis, G.M.; Harker, J.A. Innate and Adaptive Immune Regulation During Chronic Viral Infections. Annu. Rev. Virol. 2015, 2, 573–597. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Zhang, Y.; Gu, W.; Sun, B. TH1/TH2 cell differentiation and molecular signals. Adv. Exp. Med. Biol. 2014, 841, 15–44. [Google Scholar] [PubMed]
- Trinchieri, G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol. 2003, 3, 133–146. [Google Scholar] [CrossRef] [PubMed]
- Colombo, P.E.; Quenet, F.; Alric, P.; Mourregot, A.; Neron, M.; Portales, F.; Rouanet, P.; Carrier, G. Distal Pancreatectomy with Celiac Axis Resection (Modified Appleby Procedure) and Arterial Reconstruction for Locally Advanced Pancreatic Adenocarcinoma after FOLFIRINOX Chemotherapy and Chemoradiation Therapy. Ann. Surg. Oncol. 2021, 28, 1106–1108. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.; Boguniewicz, M.; Howell, M.D.; Nomura, I.; Hamid, Q.A. New insights into atopic dermatitis. J. Clin. Investig. 2004, 113, 651–657. [Google Scholar] [CrossRef] [PubMed]
- Corry, D.B.; Kheradmand, F. Induction and regulation of the IgE response. Nature 1999, 402, B18–B23. [Google Scholar] [CrossRef] [PubMed]
- Deo, S.S.; Mistry, K.J.; Kakade, A.M.; Niphadkar, P.V. Role played by Th2 type cytokines in IgE mediated allergy and asthma. Lung India 2010, 27, 66–71. [Google Scholar] [CrossRef]
- Hanson, L.A. Atopic allergies and contact allergies. In Immunology; Wigzell, L.A., Ed.; Butterworth-Heinemann: Oxford, UK, 1985; pp. 198–209. [Google Scholar] [CrossRef]
- And, K.I.; Ishizaka, T. Mechanisms of reaginic hypersensitivity and IgE antibody response. Immunol. Rev. 1978, 41, 109–148. [Google Scholar] [CrossRef]
- Hirahara, K.; Nakayama, T. CD4+ T-cell subsets in inflammatory diseases: Beyond the Th 1/Th 2 paradigm. Int. Immunol. 2016, 28, 163–171. [Google Scholar] [CrossRef] [Green Version]
- Brand, S. Crohn’s disease: Th1, Th17 or both? The change of a paradigm: New immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut 2009, 58, 1152–1167. [Google Scholar] [CrossRef] [Green Version]
- Caminati, M.; Pham, D.L.; Bagnasco, D.; Canonica, G.W. Type 2 immunity in asthma. World Allergy Organ. J. 2018, 11, 13. [Google Scholar] [CrossRef]
- Han, M.; Rajput, C.; Hong, J.Y.; Lei, J.; Hinde, J.L.; Wu, Q.; Bentley, J.K.; Hershenson, M.B. The innate cytokines IL-25, IL-33, and TSLP cooperate in the induction of type 2 innate lymphoid cell expansion and mucous metaplasia in rhinovirus-infected immature mice. J. Immunol. 2017, 199, 1308–1318. [Google Scholar] [CrossRef] [Green Version]
- Maddur, M.S.; Miossec, P.; Kaveri, S.V.; Bayry, J. Th17 Cells: Biology, Pathogenesis of Autoimmune and Inflammatory Diseases, and Therapeutic Strategies. Am. J. Pathol. 2012, 181, 8–18. [Google Scholar] [CrossRef]
- McKinley, L.; Alcorn, J.F.; Peterson, A.; DuPont, R.B.; Kapadia, S.; Logar, A.; Henry, A.; Irvin, C.G.; Piganelli, J.D.; Ray, A. TH17 cells mediate steroid-resistant airway inflammation and airway hyperresponsiveness in mice. J. Immunol. 2008, 181, 4089–4097. [Google Scholar] [CrossRef] [Green Version]
- Makris, S.; Johnston, S. Recent advances in understanding rhinovirus immunity. F1000 Res. 2018, 7, 1537. [Google Scholar] [CrossRef]
- Simbirtsev, A.S.; Kozlov, I.G. Cytokine System. In Mechanical Stretch and Cytokines; Kamkin, A., Kiseleva, I., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 1–33. [Google Scholar]
- Induction and Regulation of IFNs During Viral Infections. J. Interferon Cytokine Res. 2004, 24, 439–454. [CrossRef]
- Ank, N.; West, H.; Bartholdy, C.; Eriksson, K.; Thomsen, A.R.; Paludan, S.R. Lambda Interferon (IFN-λ), a Type III IFN, Is Induced by Viruses and IFNs and Displays Potent Antiviral Activity against Select Virus Infections In Vivo. J. Virol. 2006, 80, 4501–4509. [Google Scholar] [CrossRef] [Green Version]
- Perry, A.K.; Chen, G.; Zheng, D.; Tang, H.; Cheng, G. The host type I interferon response to viral and bacterial infections. Cell Res. 2005, 15, 407–422. [Google Scholar] [CrossRef] [Green Version]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Sada, M.; Watanabe, M.; Inui, T.; Nakamoto, K.; Hirata, A.; Nakamura, M.; Honda, K.; Saraya, T.; Kurai, D.; Kimura, H. Ruxolitinib inhibits poly (I:C) and type 2 cytokines-induced CCL5 production in bronchial epithelial cells: A potential therapeutic agent for severe eosinophilic asthma. Immun. Inflamm. Dis. 2021, 9, 363–373. [Google Scholar] [CrossRef]
- Galkina, E.; Thatte, J.; Dabak, V.; Williams, M.B.; Ley, K.; Braciale, T.J. Preferential migration of effector CD8+ T cells into the interstitium of the normal lung. J. Clin. Investig. 2005, 115, 3473–3483. [Google Scholar] [CrossRef]
- Kohlmeier, J.E.; Miller, S.C.; Smith, J.; Lu, B.; Gerard, C.; Cookenham, T.; Roberts, A.D.; Woodland, D.L. The chemokine receptor CCR5 plays a key role in the early memory CD8+ T cell response to respiratory virus infections. Immunity 2008, 29, 101–113. [Google Scholar] [CrossRef] [Green Version]
- Tekkanat, K.K.; Maassab, H.; Miller, A.; Berlin, A.A.; Kunkel, S.L.; Lukacs, N.W. RANTES (CCL5) production during primary respiratory syncytial virus infection exacerbates airway disease. Eur. J. Immunol. 2002, 32, 3276–3284. [Google Scholar] [CrossRef] [PubMed]
- Culley, F.J.; Pennycook, A.M.; Tregoning, J.S.; Dodd, J.S.; Walzl, G.; Wells, T.N.; Hussell, T.; Openshaw, P.J. Role of CCL5 (RANTES) in viral lung disease. J. Virol. 2006, 80, 8151–8157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krammer, S.; Gutu, C.S.; Grund, J.C.; Chiriac, M.T.; Zirlik, S.; Finotto, S. Regulation and Function of Interferon-Lambda (IFNλ) and Its Receptor in Asthma. Front. Immunol. 2021, 12, 731807. [Google Scholar] [CrossRef] [PubMed]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-λ Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Koch, S.; Finotto, S. Role of Interferon-λ in Allergic Asthma. J. Innate Immun. 2015, 7, 224–230. [Google Scholar] [CrossRef]
- Won, J.; Gil, C.H.; Jo, A.; Kim, H.J. Inhaled delivery of Interferon-lambda restricts epithelial-derived Th2 inflammation in allergic asthma. Cytokine 2019, 119, 32–36. [Google Scholar] [CrossRef]
- Newcomb, D.C.; Sajjan, U.S.; Nagarkar, D.R.; Goldsmith, A.M.; Bentley, J.K.; Hershenson, M.B. Cooperative effects of rhinovirus and TNF-α on airway epithelial cell chemokine expression. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2007, 293, L1021–L1028. [Google Scholar] [CrossRef]
- Liao, W.; Lin, J.-X.; Leonard, W.J. IL-2 family cytokines: New insights into the complex roles of IL-2 as a broad regulator of T helper cell differentiation. Curr. Opin. Immunol. 2011, 23, 598–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.A. GM-CSF in inflammation. J. Exp. Med. 2020, 217, e20190945. [Google Scholar] [CrossRef]
- Smiley, S.T.; Grusby, M.J. Interleukin 4. In Encyclopedia of Immunology, 2nd ed.; Delves, P.J., Ed.; Elsevier: Oxford, UK, 1998; pp. 1451–1453. [Google Scholar]
- Fulkerson, P.C.; Rothenberg, M.E. Chapter One—Eosinophil Development, Disease Involvement, and Therapeutic Suppression. In Advances in Immunology; Alt, F., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 138, pp. 1–34. [Google Scholar]
- Griffin, D.E. Cytokines and Chemokines. In Encyclopedia of VirologyI, 3rd ed.; Mahy, B.W.J., Van Regenmortel, M.H.V., Eds.; Academic Press: Oxford, UK, 2008; pp. 620–624. [Google Scholar]
- Matsuda, T.; Kishimoto, T. Interleukin 6. In Encyclopedia of Immunology, 2nd ed.; Delves, P.J., Ed.; Elsevier: Oxford, UK, 1998; pp. 1458–1461. [Google Scholar]
- Sahiner, U.; Akdis, M.; Akdis, C.A. 1—Introduction to Mechanisms of Allergic Diseases. In Allergy Essentials, 2nd ed.; O’Hehir, R.E., Holgate, S.T., Hershey, G.K.K., Sheikh, A., Eds.; Elsevier: Philadelphia, PA, USA, 2022; pp. 1–24. [Google Scholar]
- Mak, T.W.; Saunders, M.E. 17—Cytokines and Cytokine Receptors. In The Immune Response; Mak, T.W., Saunders, M.E., Eds.; Academic Press: Burlington, VT, USA, 2006; pp. 463–516. [Google Scholar]
- Ohne, Y.; Silver, J.S.; Thompson-Snipes, L.; Collet, M.A.; Blanck, J.P.; Cantarel, B.L.; Copenhaver, A.M.; Humbles, A.A.; Liu, Y.-J. IL-1 is a critical regulator of group 2 innate lymphoid cell function and plasticity. Nat. Immunol. 2016, 17, 646–655. [Google Scholar] [CrossRef]
- Jackson, D.J.; Makrinioti, H.; Rana, B.M.; Shamji, B.W.; Trujillo-Torralbo, M.-B.; Footitt, J.; Del-Rosario, J.; Telcian, A.G.; Nikonova, A.; Zhu, J. IL-33–dependent type 2 inflammation during rhinovirus-induced asthma exacerbations in vivo. Am. J. Respir. Crit. Care Med. 2014, 190, 1373–1382. [Google Scholar] [CrossRef] [Green Version]
- Burleson, S.C.M.; Fick, R.B.; Mannie, M.D.; Olmstead, S.G.; Van Scott, M.R. Chapter 35—The Immune Basis of Allergic Lung Disease. In Comparative Biology of the Normal Lung, 2nd ed.; Parent, R.A., Ed.; Academic Press: San Diego, CA, USA, 2015; pp. 683–719. [Google Scholar]
- Fujiwara, K.; Matsukawa, A.; Ohkawara, S.; Takagi, K.; Yoshinaga, M. Functional distinction between CXC chemokines, interleukin-8 (IL-8), and growth related oncogene (GRO)alpha in neutrophil infiltration. Lab. Investig. 2002, 82, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Al-Alwan, L.A.; Chang, Y.; Mogas, A.; Halayko, A.J.; Baglole, C.J.; Martin, J.G.; Rousseau, S.; Eidelman, D.H.; Hamid, Q. Differential roles of CXCL2 and CXCL3 and their receptors in regulating normal and asthmatic airway smooth muscle cell migration. J. Immunol. 2013, 191, 2731–2741. [Google Scholar] [CrossRef] [Green Version]
- Walz, A.; Schmutz, P.; Mueller, C.; Schnyder-Candrian, S. Regulation and function of the CXC chemokine ENA-78 in monocytes and its role in disease. J. Leukoc. Biol. 1997, 62, 604–611. [Google Scholar] [CrossRef]
- Mukaida, N. Pathophysiological roles of interleukin-8/CXCL8 in pulmonary diseases. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2003, 284, L566–L577. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Guo, S.; Hibbert, J.M.; Jain, V.; Singh, N.; Wilson, N.O.; Stiles, J.K. CXCL10/IP-10 in infectious diseases pathogenesis and potential therapeutic implications. Cytokine Growth Factor Rev. 2011, 22, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Anshita, D.; Ravichandiran, V. MCP-1: Function, regulation, and involvement in disease. Int. Immunopharmacol. 2021, 101, 107598. [Google Scholar] [CrossRef]
- Maurer, M.; von Stebut, E. Macrophage inflammatory protein-1. Int. J. Biochem. Cell Biol. 2004, 36, 1882–1886. [Google Scholar] [CrossRef] [PubMed]
- Rot, A.; Krieger, M.; Brunner, T.; Bischoff, S.C.; Schall, T.; Dahinden, C. RANTES and macrophage inflammatory protein 1 alpha induce the migration and activation of normal human eosinophil granulocytes. J. Exp. Med. 1992, 176, 1489–1495. [Google Scholar] [CrossRef]
- Levy, J.A. The unexpected pleiotropic activities of RANTES. J. Immunol. 2009, 182, 3945–3946. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Zepeda, E.A.; Rothenberg, M.E.; Ownbey, R.T.; Celestin, J.; Leder, P.; Luster, A.D. Human eotaxin is a specific chemoattractant for eosinophil cells and provides a new mechanism to explain tissue eosinophilia. Nat. Med. 1996, 2, 449–456. [Google Scholar] [CrossRef]
- Le Borgne, M.; Etchart, N.; Goubier, A.; Lira, S.A.; Sirard, J.C.; van Rooijen, N.; Caux, C.; Aït-Yahia, S.; Vicari, A.; Kaiserlian, D.; et al. Dendritic cells rapidly recruited into epithelial tissues via CCR6/CCL20 are responsible for CD8+ T cell crosspriming in vivo. Immunity 2006, 24, 191–201. [Google Scholar] [CrossRef]
- Roan, F.; Bell, B.D.; Stoklasek, T.A.; Kitajima, M.; Han, H.; Ziegler, S.F. The multiple facets of thymic stromal lymphopoietin (TSLP) during allergic inflammation and beyond. J. Leukoc. Biol. 2012, 91, 877–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaidi, M.R.; Merlino, G. The Two Faces of Interferon-γ in Cancer. Clin. Cancer Res. 2011, 17, 6118–6124. [Google Scholar] [CrossRef] [Green Version]
- Atzeni, F.; Sarzi-Puttini, P. Tumor Necrosis Factor. In Brenner’s Encyclopedia of Genetics, 2nd ed.; Maloy, S., Hughes, K., Eds.; Academic Press: San Diego, CA, USA, 2013; pp. 229–231. [Google Scholar]
- Tumor necrosis factor alfa. In Meyler’s Side Effects of Drugs: The International Encyclopedia of Adverse Drug Reactions and Interactions, 15th ed.; Aronson, J.K. (Ed.) Elsevier: Amsterdam, The Netherlands, 2006; pp. 3537–3538. [Google Scholar]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar] [PubMed]
- Butcher, M.J.; Zhu, J. Recent advances in understanding the Th1/Th2 effector choice. Fac. Rev. 2021, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.M.-Y.; Yip, T.-F.; Yan, S.; Jin, D.-Y.; Wei, H.-L.; Guo, R.-T.; Peiris, J.S.M. Recognition of double-stranded RNA and regulation of interferon pathway by toll-like receptor 10. Front. Immunol. 2018, 9, 516. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, M.; Seya, T. TLR3: Interferon induction by double-stranded RNA including poly(I:C). Adv. Drug Deliv. Rev. 2008, 60, 805–812. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Zhang, J.; Yu, F.S. Toll-like receptor 3 agonist poly(I:C)-induced antiviral response in human corneal epithelial cells. Immunology 2006, 117, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, K.; Watanabe, M.; Sada, M.; Inui, T.; Nakamura, M.; Honda, K.; Wada, H.; Ishii, H.; Takizawa, H. Pseudomonas aeruginosa-derived flagellin stimulates IL-6 and IL-8 production in human bronchial epithelial cells: A potential mechanism for progression and exacerbation of COPD. Exp. Lung Res. 2019, 45, 255–266. [Google Scholar] [CrossRef]
- Michalik, M.; Wójcik-Pszczoła, K.; Paw, M.; Wnuk, D.; Koczurkiewicz, P.; Sanak, M.; Pękala, E.; Madeja, Z. Fibroblast-to-myofibroblast transition in bronchial asthma. Cell. Mol. Life Sci. 2018, 75, 3943–3961. [Google Scholar] [CrossRef] [Green Version]
- Gomes, I.; Mathur, S.K.; Espenshade, B.M.; Mori, Y.; Varga, J.; Ackerman, S.J. Eosinophil-fibroblast interactions induce fibroblast IL-6 secretion and extracellular matrix gene expression: Implications in fibrogenesis. J. Allergy Clin. Immunol. 2005, 116, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Nagarkar, D.R.; Bowman, E.R.; Schneider, D.; Gosangi, B.; Lei, J.; Zhao, Y.; McHenry, C.L.; Burgens, R.V.; Miller, D.J. Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses. J. Immunol. 2009, 183, 6989–6997. [Google Scholar] [CrossRef] [Green Version]
- Kato, A.; Favoreto, S.; Avila, P.C.; Schleimer, R.P. TLR3-and Th2 cytokine-dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J. Immunol. 2007, 179, 1080–1087. [Google Scholar] [CrossRef]
- Hewson, C.A.; Jardine, A.; Edwards, M.R.; Laza-Stanca, V.; Johnston, S.L. Toll-like receptor 3 is induced by and mediates antiviral activity against rhinovirus infection of human bronchial epithelial cells. J. Virol. 2005, 79, 12273–12279. [Google Scholar] [CrossRef] [Green Version]
- Xagorari, A.; Chlichlia, K. Toll-like receptors and viruses: Induction of innate antiviral immune responses. Open Microbiol. J. 2008, 2, 49. [Google Scholar] [CrossRef] [PubMed]
- Caron, G.; Duluc, D.; Frémaux, I.; Jeannin, P.; David, C.; Gascan, H.; Delneste, Y. Direct stimulation of human T cells via TLR5 and TLR7/8: Flagellin and R-848 up-regulate proliferation and IFN-γ production by memory CD4+ T cells. J. Immunol. 2005, 175, 1551–1557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsukura, S.; Kokubu, F.; Kurokawa, M.; Kawaguchi, M.; Ieki, K.; Kuga, H.; Odaka, M.; Suzuki, S.; Watanabe, S.; Homma, T. Role of RIG-I, MDA-5, and PKR on the expression of inflammatory chemokines induced by synthetic dsRNA in airway epithelial cells. Int. Arch. Allergy Immunol. 2007, 143, 80–83. [Google Scholar] [CrossRef]
- Cardinale, F.; Giordano, P.; Chinellato, I.; Tesse, R. Respiratory epithelial imbalances in asthma pathophysiology. Allergy Asthma Proc. 2013, 34, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Comstock, A.T.; Ganesan, S.; Chattoraj, A.; Faris, A.N.; Margolis, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J. Virol. 2011, 85, 6795–6808. [Google Scholar] [CrossRef] [Green Version]
- Rajan, D.; McCracken, C.E.; Kopleman, H.B.; Kyu, S.Y.; Lee, F.E.-H.; Lu, X.; Anderson, L.J. Human rhinovirus induced cytokine/chemokine responses in human airway epithelial and immune cells. PLoS ONE 2014, 9, e114322. [Google Scholar] [CrossRef] [Green Version]
- Ghildyal, R.; Dagher, H.; Donninger, H.; de Silva, D.; Li, X.; Freezer, N.J.; Wilson, J.W.; Bardin, P.G. Rhinovirus infects primary human airway fibroblasts and induces a neutrophil chemokine and a permeability factor. J. Med. Virol. 2005, 75, 608–615. [Google Scholar] [CrossRef]
- Hayden, F.G. Rhinovirus and the lower respiratory tract. Rev. Med. Virol. 2004, 14, 17–31. [Google Scholar] [CrossRef]
- Muehling, L.M.; Heymann, P.W.; Wright, P.W.; Eccles, J.D.; Agrawal, R.; Carper, H.T.; Murphy, D.D.; Workman, L.J.; Word, C.R.; Ratcliffe, S.J.; et al. Human TH1 and TH2 cells targeting rhinovirus and allergen coordinately promote allergic asthma. J. Allergy Clin. Immunol. 2020, 146, 555–570. [Google Scholar] [CrossRef] [PubMed]
- Gern, J.E.; Vrtis, R.; Grindle, K.A.; Swenson, C.; Busse, W.W. Relationship of upper and lower airway cytokines to outcome of experimental rhinovirus infection. Am. J. Respir. Crit. Care Med. 2000, 162, 2226–2231. [Google Scholar] [CrossRef]
- Thomas, B.J.; Lindsay, M.; Dagher, H.; Freezer, N.J.; Li, D.; Ghildyal, R.; Bardin, P.G. Transforming growth factor-beta enhances rhinovirus infection by diminishing early innate responses. Am. J. Respir. Cell Mol. Biol. 2009, 41, 339–347. [Google Scholar] [CrossRef] [PubMed]
- O’Garra, A. Cytokines induce the development of functionally heterogeneous T helper cell subsets. Immunity 1998, 8, 275–283. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.M.; An, J. Cytokines, inflammation, and pain. Int. Anesthesiol. Clin. 2007, 45, 27–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gern, J.E.; Busse, W.W. Association of rhinovirus infections with asthma. Clin. Microbiol. Rev. 1999, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Palomino, D.C.; Marti, L.C. Chemokines and immunity. Einstein 2015, 13, 469–473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Juremalm, M.; Nilsson, G. Chemokine receptor expression by mast cells. Chem. Immunol. Allergy 2005, 87, 130–144. [Google Scholar] [CrossRef]
- Sheeran, P.; Jafri, H.; Carubelli, C.; Saavedra, J.; Johnson, C.; Krisher, K.; Sánchez, P.J.; Ramilo, O. Elevated cytokine concentrations in the nasopharyngeal and tracheal secretions of children with respiratory syncytial virus disease. Pediatr. Infect. Dis. J. 1999, 18, 115–122. [Google Scholar] [CrossRef]
- Folkerts, G.; Busse, W.W.; Nijkamp, F.P.; Sorkness, R.; Gern, J.E. Virus-induced airway hyperresponsiveness and asthma. Am. J. Respir. Crit. Care Med. 1998, 157, 1708–1720. [Google Scholar] [CrossRef] [PubMed]
- van Erp, E.A.; Luytjes, W.; Ferwerda, G.; van Kasteren, P.B. Fc-Mediated Antibody Effector Functions during Respiratory Syncytial Virus Infection and Disease. Front. Immunol. 2019, 10, 548. [Google Scholar] [CrossRef] [Green Version]
- Crotty, S. Follicular helper CD4 T cells (Tfh). Annu. Rev. Immunol. 2011, 29, 621–663. [Google Scholar] [CrossRef]
- Aderem, A.; Underhill, D.M. Mechanisms of phagocytosis in macrophages. Annu. Rev. Immunol. 1999, 17, 593–623. [Google Scholar] [CrossRef]
- Johnston, S.L. Innate immunity in the pathogenesis of virus-induced asthma exacerbations. Proc. Am. Thorac. Soc. 2007, 4, 267–270. [Google Scholar] [CrossRef] [PubMed]
- Wark, P.A.; Gibson, P.G. Asthma exacerbations 3: Pathogenesis. Thorax 2006, 61, 909–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikhail, I.; Grayson, M.H. Asthma and viral infections: An intricate relationship. Ann. Allergy Asthma Immunol. 2019, 123, 352–358. [Google Scholar] [CrossRef]
- Barnes, P.J. Th2 cytokines and asthma: An introduction. Respir. Res. 2001, 2, 64. [Google Scholar] [CrossRef] [PubMed]
- Brandt, E.B.; Sivaprasad, U. Th2 Cytokines and Atopic Dermatitis. J. Clin. Cell. Immunol. 2011, 2, 1000110. [Google Scholar] [CrossRef] [PubMed]
- Kay, A.B.; Phipps, S.; Robinson, D.S. A role for eosinophils in airway remodelling in asthma. Trends Immunol. 2004, 25, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Chihara, J. Eosinophil activation by eotaxin—Eotaxin primes the production of reactive oxygen species from eosinophils. Allergy 1999, 54, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Herbert, C.; Zeng, Q.X.; Shanmugasundaram, R.; Garthwaite, L.; Oliver, B.G.; Kumar, R.K. Response of airway epithelial cells to double-stranded RNA in an allergic environment. Transl. Respir. Med. 2014, 2, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Djukanović, R.; Harrison, T.; Johnston, S.L.; Gabbay, F.; Wark, P.; Thomson, N.C.; Niven, R.; Singh, D.; Reddel, H.K.; Davies, D.E. The effect of inhaled IFN-β on worsening of asthma symptoms caused by viral infections. A randomized trial. Am. J. Respir. Crit. Care Med. 2014, 190, 145–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wos, M.; Sanak, M.; Soja, J.; Olechnowicz, H.; Busse, W.W.; Szczeklik, A. The presence of rhinovirus in lower airways of patients with bronchial asthma. Am. J. Respir. Crit. Care Med. 2008, 177, 1082–1089. [Google Scholar] [CrossRef] [Green Version]
- Dharmage, S.C.; Perret, J.L.; Custovic, A. Epidemiology of asthma in children and adults. Front. Pediatr. 2019, 7, 246. [Google Scholar] [CrossRef]


{kind=link}
| Cytokines | Origin | Function | Ref. No. | |
|---|---|---|---|---|
| Th1 | IFN-γ | Activated T cell | Macrophage activation | [55,74] |
| IFN-λ | Activated T cell | Dendritic cell activation | [75] | |
| TNF-α | Activated T cell, Monocyte/Macrophage | Promotes inflammatory cytokines | [50,76] | |
| IL-2 | Activated T cell | T cell/NK cell activation | [51] | |
| GM-CSF | Activated T cell Monocyte/Macrophage, Fibroblast | Neutrophil/Macrophage activation | [52] | |
| Th2 | IL-4 | Activated T cell | B cell/T cell activation IgE isotype switch | [53,58] |
| IL-5 | Activated T cell Eosinophil, Mast cell | Eosinophil proliferation/differentiation | [54,55] | |
| IL-6 | Activated T cell | Induce B cells into antibody-producing cells T-cell differentiation | [56] | |
| IL-13 | Activated T cell Mast cell | Macrophage activation IgE isotype switch | [57,58] | |
| Others | IL-1 | Monocyte/MΦ | T cell activation | [59] |
| IL-33 | Activated T cell Endothelial/Epithelial cell | Promoting Th2-associated cytokines | [60,61] | |
| CXCL1 GROα | Epithelial cells, Macrophage, Neutrophil | Neutrophil migration | [62] | |
| CXCL3 GROγ MIP-2β | Monocyte, Fibroblast | Neutrophil migration/adhesion | [63] | |
| CXCL5 ENA-78 | Eosinophil | Neutrophil migration/activation | [64] | |
| CXCL8 IL-8 | Monocyte/Macrophage, Fibroblast | Neutrophil migration | [62,65] | |
| CXCL10 IP-10 | Monocyte, Fibroblast | T cell/NK cell activation | [66] | |
| CCL2 MCP-1 | Monocyte/Macrophage, Fibroblast | Monocyte migration | [67] | |
| CCL3 MIP-1α | Monocyte/Macrophage, Activated T cell | Monocyte migration/infiltration Eosinophil/mast cell/Dendritic cell migration | [68] | |
| CCL4 MIP-1β | Monocyte/Macrophage, Activated T cell | Monocyte/T cell migration | [68] | |
| CCL5 RANTES | Monocyte/Macrophage, Fibroblast | Monocyte/lymphocyte/Eosinophil/mast cell migration | [69,70] | |
| CCL11 Eotaxin | Airway Epithelial Cells, Fibroblast | Eosinophil migration/degranulation Mast cell activation/migration | [71] | |
| CCL20 MIP-3α | Neutrophil, Natural Killer Cell | Dendritic cell activation/migration T cell/B cell migration | [72] | |
| CCL24 Eotaxin-2 | Monocyte/Macrophage | Eosinophil migration | [71] | |
| TSLP | Epithelial cells | Promoting Th2-associated cytokines B cell activation | [73] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hayashi, Y.; Sada, M.; Shirai, T.; Okayama, K.; Kimura, R.; Kondo, M.; Okodo, M.; Tsugawa, T.; Ryo, A.; Kimura, H. Rhinovirus Infection and Virus-Induced Asthma. Viruses 2022, 14, 2616. https://doi.org/10.3390/v14122616
Hayashi Y, Sada M, Shirai T, Okayama K, Kimura R, Kondo M, Okodo M, Tsugawa T, Ryo A, Kimura H. Rhinovirus Infection and Virus-Induced Asthma. Viruses. 2022; 14(12):2616. https://doi.org/10.3390/v14122616
Chicago/Turabian StyleHayashi, Yuriko, Mitsuru Sada, Tatsuya Shirai, Kaori Okayama, Ryusuke Kimura, Mayumi Kondo, Mitsuaki Okodo, Takeshi Tsugawa, Akihide Ryo, and Hirokazu Kimura. 2022. "Rhinovirus Infection and Virus-Induced Asthma" Viruses 14, no. 12: 2616. https://doi.org/10.3390/v14122616
APA StyleHayashi, Y., Sada, M., Shirai, T., Okayama, K., Kimura, R., Kondo, M., Okodo, M., Tsugawa, T., Ryo, A., & Kimura, H. (2022). Rhinovirus Infection and Virus-Induced Asthma. Viruses, 14(12), 2616. https://doi.org/10.3390/v14122616