
Targeting a Conserved Lysine in the Hydrophobic Pocket of HIV-1 gp41 Improves Small Molecule Antiviral Activity

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
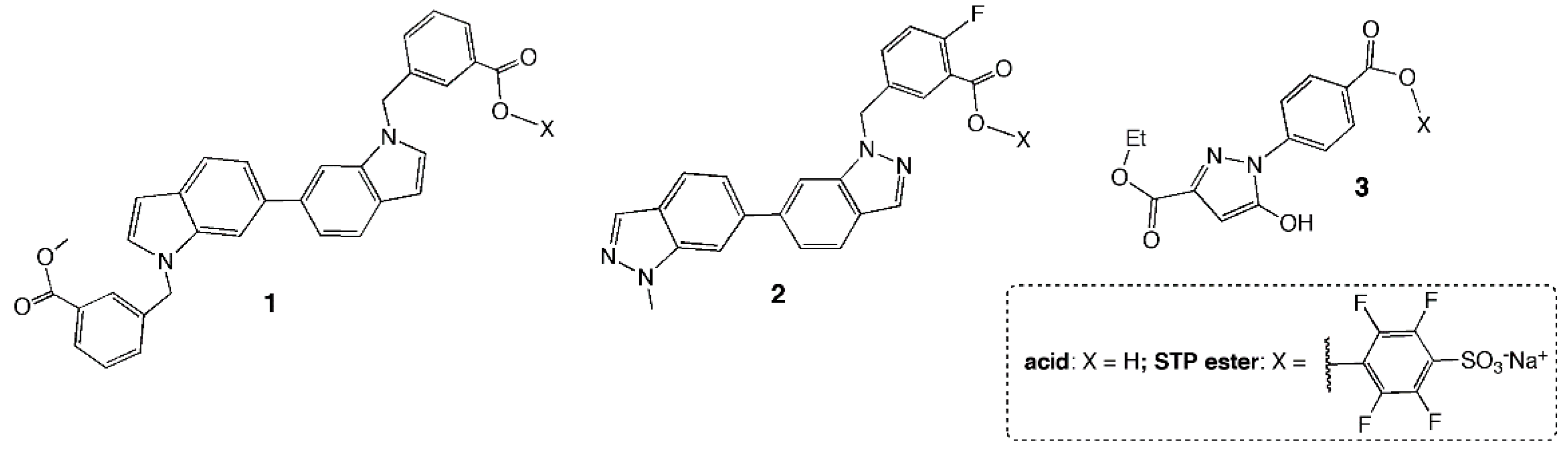
2.1. Chemistry
2.2. Binding Affinity Assay
2.3. Protein Constructs
2.4. Cell-Based Assays
3. Results
3.1. STP Esters Readily Reacted with Free Lysine-ε-NH2
3.2. STP Esterification of 2 and 3 Improved Binding Affinity for the HP
3.3. Compounds 2-STP and 3-STP Reacted with a Protein Construct of the gp41 Ectodomain
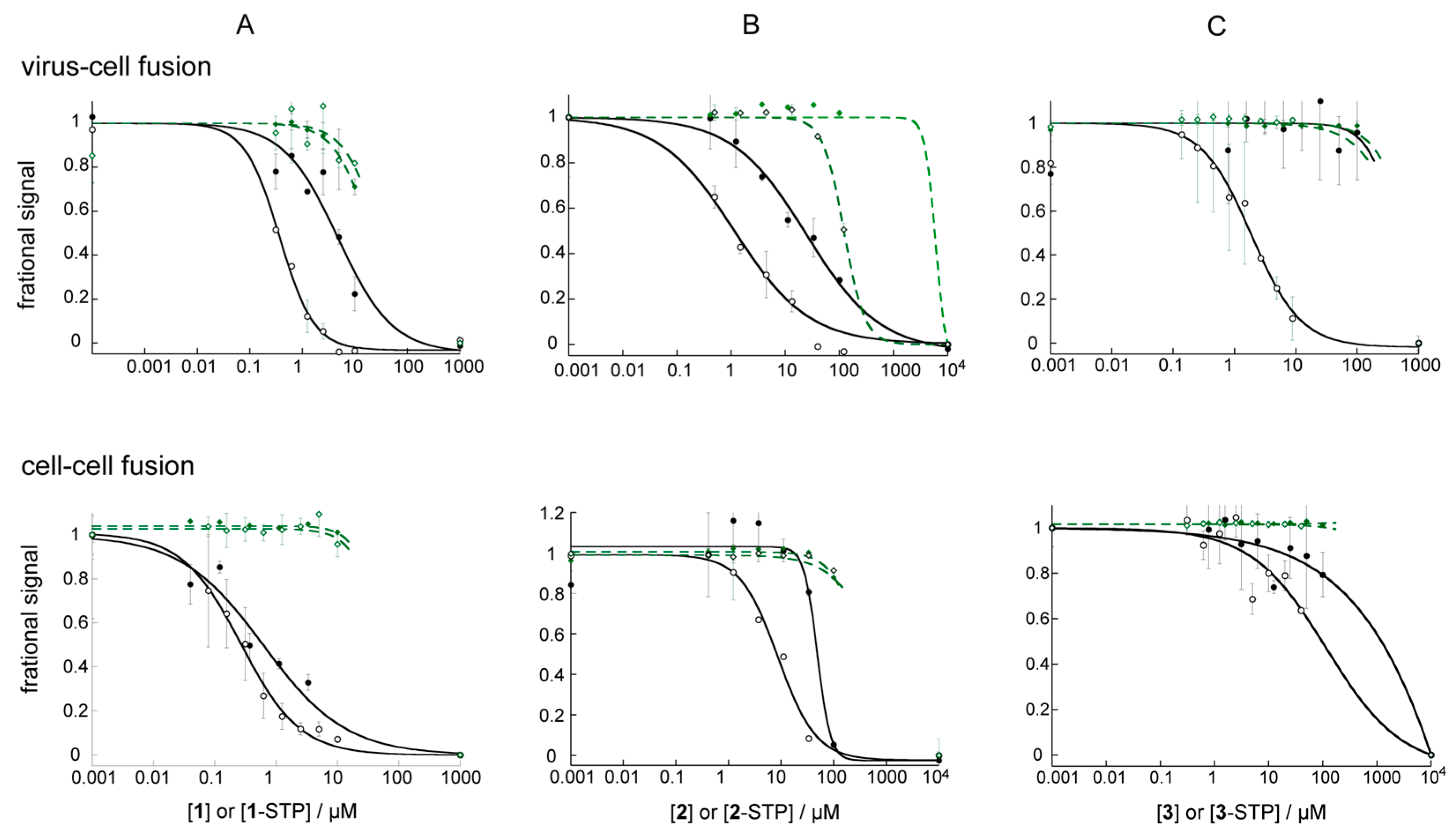
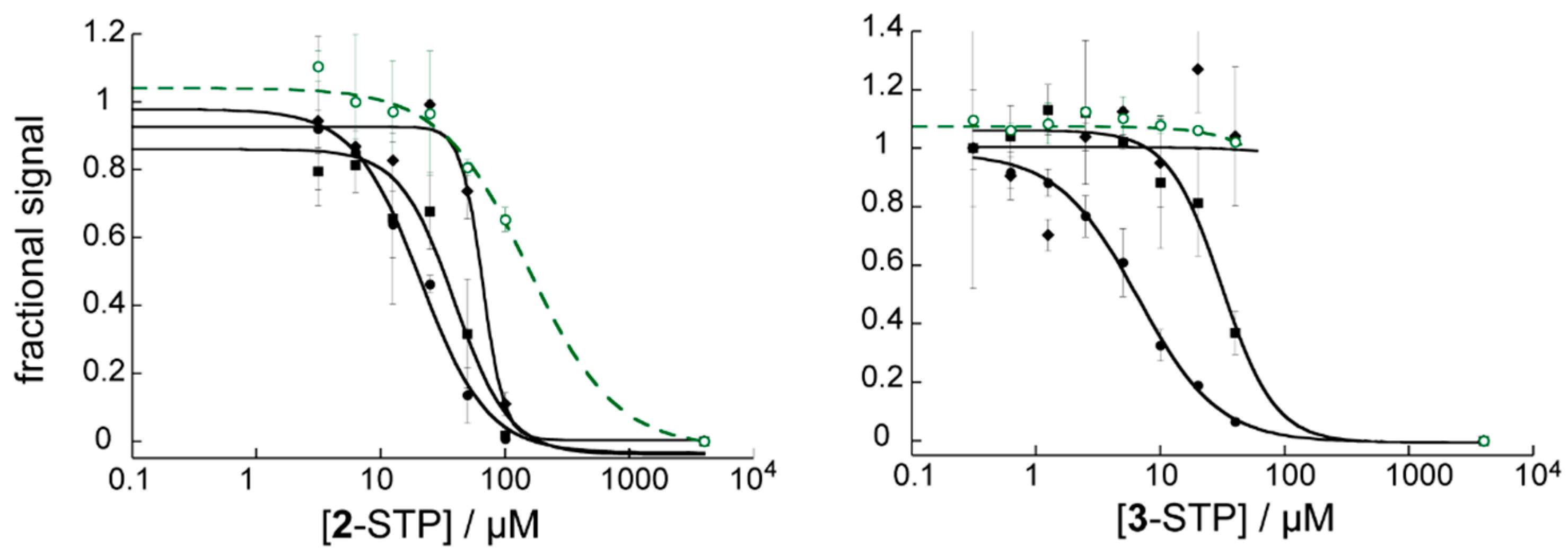
3.4. STP Esterification Significantly Improved the Antiviral Activity of the Compounds
3.5. The Effect of STP Esterification on Efficacy against Cell–Cell Fusion Was Not as Clear
3.6. STP Esters Act Specifically on HIV Env Mediated Fusion
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef] [Green Version]
- Tan, K.; Liu, J.; Wang, J.; Shen, S.; Lu, M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. USA 1997, 94, 12303–12308. [Google Scholar] [CrossRef] [Green Version]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41 [see comments]. Nature 1997, 387, 426–430. [Google Scholar] [CrossRef]
- Melikyan, G.B.; Markosyan, R.M.; Hemmati, H.; Delmedico, M.K.; Lambert, D.M.; Cohen, F.S. Evidence that the transition of HIV-1 gp41 into a six-helix bundle, not the bundle configuration, induces membrane fusion. J. Cell Biol. 2000, 151, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Chan, D.C.; Chutkowski, C.T.; Kim, P.S. Evidence that a prominent cavity in the coiled coil of HIV type 1 gp41 is an attractive drug target. Proc. Natl. Acad. Sci. USA 1998, 95, 15613–15617. [Google Scholar] [CrossRef] [Green Version]
- Strockbine, B.; Rizzo, R.C. Binding of antifusion peptides with HIVgp41 from molecular dynamics simulations: Quantitative correlation with experiment. Proteins 2007, 67, 630–642. [Google Scholar] [CrossRef]
- Valades-Alcaraz, A.; Reinosa, R.; Holguin, A. HIV Transmembrane Glycoprotein Conserved Domains and Genetic Markers Across HIV-1 and HIV-2 Variants. Front. Microbiol. 2022, 13, 855232. [Google Scholar] [CrossRef] [PubMed]
- Su, Y.; Chong, H.; Qiu, Z.; Xiong, S.; He, Y. Mechanism of HIV-1 Resistance to Short-Peptide Fusion Inhibitors Targeting the Gp41 Pocket. J. Virol. 2015, 89, 5801–5811. [Google Scholar] [CrossRef] [Green Version]
- Smith, A.R.; Weinstock, M.T.; Siglin, A.E.; Whitby, F.G.; Francis, J.N.; Hill, C.P.; Eckert, D.M.; Root, M.J.; Kay, M.S. Characterization of resistance to a potent D-peptide HIV entry inhibitor. Retrovirology 2019, 16, 28. [Google Scholar] [CrossRef] [Green Version]
- Lu, K.; Asyifah, M.R.; Shao, F.; Zhang, D. Development of HIV-1 fusion inhibitors targeting gp41. Curr. Med. Chem. 2014, 21, 1976–1996. [Google Scholar] [CrossRef]
- Lu, L.; Yu, F.; Cai, L.; Debnath, A.K.; Jiang, S. Development of Small-molecule HIV Entry Inhibitors Specifically Targeting gp120 or gp41. Curr. Top. Med. Chem. 2016, 16, 1074–1090. [Google Scholar] [CrossRef] [Green Version]
- Qiu, J.; Liang, T.; Wu, J.; Yu, F.; He, X.; Tian, Y.; Xie, L.; Jiang, S.; Liu, S.; Li, L. N-Substituted Pyrrole Derivative 12m Inhibits HIV-1 Entry by Targeting Gp41 of HIV-1 Envelope Glycoprotein. Front. Pharmacol. 2019, 10, 859. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Chu, S.; Nemati, A.; Huang, C.; Snyder, B.A.; Ptak, R.G.; Gochin, M. Investigation of the molecular characteristics of bisindole inhibitors as HIV-1 glycoprotein-41 fusion inhibitors. Eur. J. Med. Chem. 2019, 161, 533–542. [Google Scholar] [CrossRef]
- Zhou, G.; Sofiyev, V.; Kaur, H.; Snyder, B.A.; Mankowski, M.K.; Hogan, P.A.; Ptak, R.G.; Gochin, M. Structure-Activity Relationship Studies of Indole-Based Compounds as Small Molecule HIV-1 Fusion Inhibitors Targeting Glycoprotein 41. J. Med. Chem. 2014, 57, 5270–5281. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Wu, D.; Snyder, B.; Ptak, R.G.; Kaur, H.; Gochin, M. Development of indole compounds as small molecule inhibitors of HIV-1 gp41. J. Med. Chem. 2011, 54, 7220–7231. [Google Scholar] [CrossRef] [Green Version]
- Baillie, T.A. Targeted Covalent Inhibitors for Drug Design. Angew. Chem. Int. Ed. 2016, 55, 13408–13421. (In English) [Google Scholar] [CrossRef]
- Gambini, L.; Baggio, C.; Udompholkul, P.; Jossart, J.; Salem, A.F.; Perry, J.J.P.; Pellecchia, M. Covalent Inhibitors of Protein-Protein Interactions Targeting Lysine, Tyrosine, or Histidine Residues. J. Med. Chem. 2019, 62, 5616–5627. [Google Scholar] [CrossRef] [Green Version]
- Cuesta, A.; Taunton, J. Lysine-Targeted Inhibitors and Chemoproteomic Probes. Annu. Rev. Biochem. 2019, 88, 365–381. [Google Scholar] [CrossRef]
- Zhou, G.; He, L.; Li, K.H.; Pedroso, C.C.S.; Gochin, M. A targeted covalent small molecule inhibitor of HIV-1 fusion. Chem. Commun. (Camb.) 2021, 57, 4528–4531. [Google Scholar] [CrossRef]
- Pettinger, J.; Jones, K.; Cheeseman, M.D. Lysine-Targeting Covalent Inhibitors. Angew. Chem. Int. Ed. 2017, 56, 15200–15209. (In English) [Google Scholar] [CrossRef]
- Cai, L.; Gochin, M. A Novel Fluorescence Intensity Screening Assay Identifies New Low Molecular Weight Inhibitors of the gp41 Coiled Coil Domain of HIV-1. Antimicrob. Agents Chemother. 2007, 51, 2388–2395. [Google Scholar] [CrossRef] [Green Version]
- Gochin, M. A suite of modular fluorescence assays interrogate the human immunodeficiency virus glycoprotein-41 coiled coil and assist in determining binding mechanism of low molecular weight fusion inhibitors. Assay Drug Dev. Technol. 2012, 10, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.D.; Chu, S.; Zhang, S.Q.; Gochin, M. Design and characterization of swapped-domain constructs of HIV-1 glycoprotein-41 as receptors for drug discovery. Protein Eng. Des. Sel. 2015, 28, 107–116. [Google Scholar] [CrossRef]
- Platt, E.J.; Wehrly, K.; Kuhmann, S.E.; Chesebro, B.; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol. 1998, 72, 2855–2864. [Google Scholar] [CrossRef] [Green Version]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [Green Version]
- Ciminale, V.; Felber, B.K.; Campbell, M.; Pavlakis, G.N. A bioassay for HIV-1 based on Env-CD4 interaction. AIDS Res. Hum. Retrovir. 1990, 6, 1281–1287. [Google Scholar] [CrossRef]
- Li, M.; Gao, F.; Mascola, J.R.; Stamatatos, L.; Polonis, V.R.; Koutsoukos, M.; Voss, G.; Goepfert, P.; Gilbert, P.; Greene, K.M.; et al. Human immunodeficiency virus type 1 env clones from acute and early subtype B infections for standardized assessments of vaccine-elicited neutralizing antibodies. J. Virol. 2005, 79, 10108–10125. [Google Scholar] [CrossRef] [Green Version]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef]
- Melikyan, G.B. HIV entry: A game of hide-and-fuse? Curr. Opin. Virol. 2014, 4, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Permanyer, M.; Ballana, E.; Este, J.A. Endocytosis of HIV: Anything goes. Trends Microbiol. 2010, 18, 543–551. [Google Scholar] [CrossRef]
- Miyauchi, K.; Kozlov, M.M.; Melikyan, G.B. Early steps of HIV-1 fusion define the sensitivity to inhibitory peptides that block 6-helix bundle formation. PLoS Pathog. 2009, 5, e1000585. [Google Scholar] [CrossRef] [Green Version]
- Daelemans, D.; Pauwels, R.; De Clercq, E.; Pannecouque, C. A time-of-drug addition approach to target identification of antiviral compounds. Nat. Protoc. 2011, 6, 925–933. [Google Scholar] [CrossRef]
- Rusconi, S.; Scozzafava, A.; Mastrolorenzo, A.; Supuran, C.T. An Update in the Development of HIV Entry Inhibitors. Curr. Top. Med. Chem. 2007, 7, 1273–1289. [Google Scholar] [CrossRef]
- Hacker, S.M.; Backus, K.M.; Lazear, M.R.; Forli, S.; Correia, B.E.; Cravatt, B.F. Global profiling of lysine reactivity and ligandability in the human proteome. Nat. Chem. 2017, 9, 1181–1190. [Google Scholar] [CrossRef]
- He, X.Y.; Lu, L.; Qiu, J.; Zou, P.; Yu, F.; Jiang, X.K.; Li, L.; Jiang, S.; Liu, S.; Xie, L. Small molecule fusion inhibitors: Design, synthesis and biological evaluation of (Z)-3-(5-(3-benzyl-4-oxo-2-thioxothiazolidinylidene)methyl)-N-(3-carboxy-4-hydrox y)phenyl-2,5-dimethylpyrroles and related derivatives targeting HIV-1 gp41. Bioorganic Med. Chem. 2013, 21, 7539–7548. [Google Scholar] [CrossRef]
- Jiang, S.; Tala, S.R.; Lu, H.; Abo-Dya, N.E.; Avan, I.; Gyanda, K.; Lu, L.; Katritzky, A.R.; Debnath, A.K. Design, Synthesis, and Biological Activity of Novel 5-((Arylfuran/1H-pyrrol-2-yl)methylene)-2-thioxo-3-(3-(trifluoromethyl)phe nyl)thiazolidin-4-ones as HIV-1 Fusion Inhibitors Targeting gp41. J. Med. Chem. 2011, 54, 572–579. [Google Scholar] [CrossRef]
- Welch, B.D.; Francis, J.N.; Redman, J.S.; Paul, S.; Weinstock, M.T.; Reeves, J.D.; Lie, Y.S.; Whitby, F.G.; Eckert, D.M.; Hill, C.P.; et al. Design of a potent D-peptide HIV-1 entry inhibitor with a strong barrier to resistance. J. Virol. 2010, 84, 11235–11244. [Google Scholar] [CrossRef] [Green Version]
- Jurado, S.; Moog, C.; Cano-Munoz, M.; Schmidt, S.; Laumond, G.; Ruocco, V.; Standoli, S.; Polo-Megias, D.; Conejero-Lara, F.; Morel, B. Probing Vulnerability of the gp41 C-Terminal Heptad Repeat as Target for Miniprotein HIV Inhibitors. J. Mol. Biol. 2020, 432, 5577–5592. [Google Scholar] [CrossRef]
- Chong, H.; Yao, X.; Qiu, Z.; Qin, B.; Han, R.; Waltersperger, S.; Wang, M.; Cui, S.; He, Y. Discovery of critical residues for viral entry and inhibition through structural Insight of HIV-1 fusion inhibitor CP621-652. J. Biol. Chem. 2012, 287, 20281–20289. [Google Scholar] [CrossRef] [Green Version]
- Chu, S.; Gochin, M. Identification of fragments targeting an alternative pocket on HIV-1 gp41 by NMR screening and similarity searching. Bioorganic Med. Chem. Lett. 2013, 23, 5114–5118. [Google Scholar] [CrossRef]
- Guan, S.; Price, J.C.; Prusiner, S.B.; Ghaemmaghami, S.; Burlingame, A.L. A data processing pipeline for mammalian proteome dynamics studies using stable isotope metabolic labeling. Mol. Cell Proteom. 2011, 10, M111.010728. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MWt | Log P † | KI a | IC50 HXB2 a,b | IC50 (CCF) a,c |
|---|---|---|---|---|---|
| 1 * | 514.57 | 7.38 | 1 ± 0.5 | 3 | 0.4 |
| 2 | 400.41 | 4.04 | 25 ± 5 | 35 | ≥50 |
| 3 | 276.24 | 1.10 | 177 ± 12 | >100 | >100 |
| Antiviral Activity | Inhibition of Cell–Cell Fusion | |||||
|---|---|---|---|---|---|---|
| Compound | EC50 | EC90 † | CC50 ‡ | EC50 | EC90 † | CC50 ‡ |
| 1 | 3.2 ± 1.6 | 17 ± 13 | 22 ± 3 | 0.4 ± 0.2 | 12 ± 6 | >>10 |
| 2 | 35 ± 7 | 800 ± 190 | ≥356 | 78 ± 24 a | 110 ± 19 a | ≥113 |
| 3 | >100 | n/a | >100 | >100 | n/a | >100 |
| 1-STP | 0.3 ± 0.2 | 2.9 ± 1.6 | 20 ± 3 | 0.23 ± 0.16 | 4.3 ± 2.8 | >>10 |
| 2-STP | 5 ± 4 | 59 ± 22 | ≥100 | 12 ± 3 | 38 ± 11 | ≥200 |
| 3-STP | 9 ± 2 | 44 ± 15 | >>40 | >40 | n/a | >>40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, L.; Zhou, G.; Sofiyev, V.; Garcia, E.; Nguyen, N.; Li, K.H.; Gochin, M. Targeting a Conserved Lysine in the Hydrophobic Pocket of HIV-1 gp41 Improves Small Molecule Antiviral Activity. Viruses 2022, 14, 2703. https://doi.org/10.3390/v14122703
He L, Zhou G, Sofiyev V, Garcia E, Nguyen N, Li KH, Gochin M. Targeting a Conserved Lysine in the Hydrophobic Pocket of HIV-1 gp41 Improves Small Molecule Antiviral Activity. Viruses. 2022; 14(12):2703. https://doi.org/10.3390/v14122703
Chicago/Turabian StyleHe, Li, Guangyan Zhou, Vladimir Sofiyev, Eddie Garcia, Newton Nguyen, Kathy H. Li, and Miriam Gochin. 2022. "Targeting a Conserved Lysine in the Hydrophobic Pocket of HIV-1 gp41 Improves Small Molecule Antiviral Activity" Viruses 14, no. 12: 2703. https://doi.org/10.3390/v14122703
APA StyleHe, L., Zhou, G., Sofiyev, V., Garcia, E., Nguyen, N., Li, K. H., & Gochin, M. (2022). Targeting a Conserved Lysine in the Hydrophobic Pocket of HIV-1 gp41 Improves Small Molecule Antiviral Activity. Viruses, 14(12), 2703. https://doi.org/10.3390/v14122703