
Enhanced Arbovirus Surveillance with High-Throughput Metatranscriptomic Processing of Field-Collected Mosquitoes
 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mosquito Collection and Initial Arbovirus Screening
2.2. Sample Preparation and Nucleic Acid Extraction
2.3. Metatranscriptomic Sequencing
2.4. Read Assembly and Taxonomic Classification
2.5. Metatranscriptomic Mosquito and Biting Midge Species Identification
2.6. Targeted Arbovirus Screen
2.7. Confirmation of Arbovirus Detections Using RT-qPCR
2.8. Phylogenetic Analysis of Target Arboviruses
2.9. Virome Analysis
3. Results
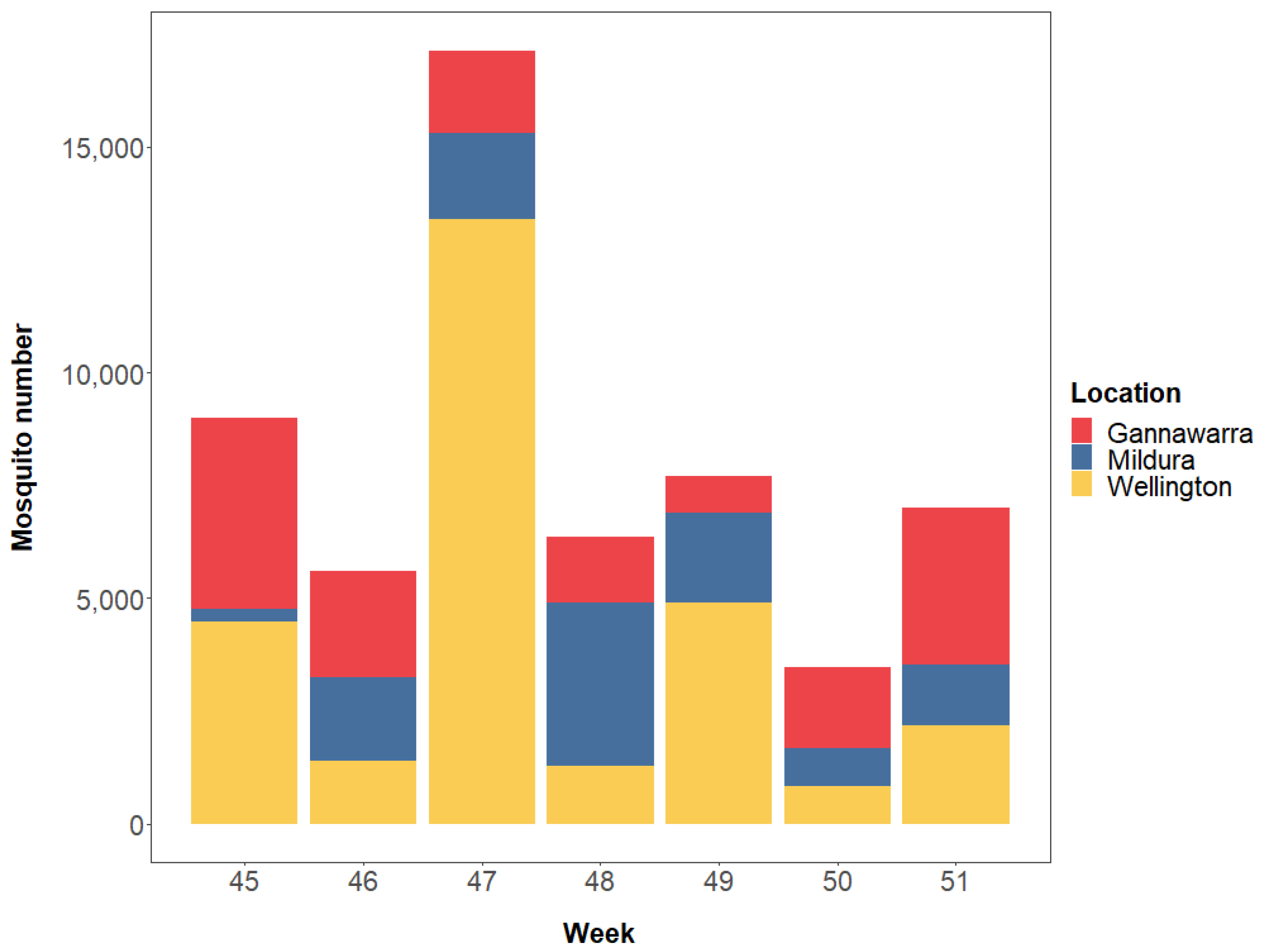
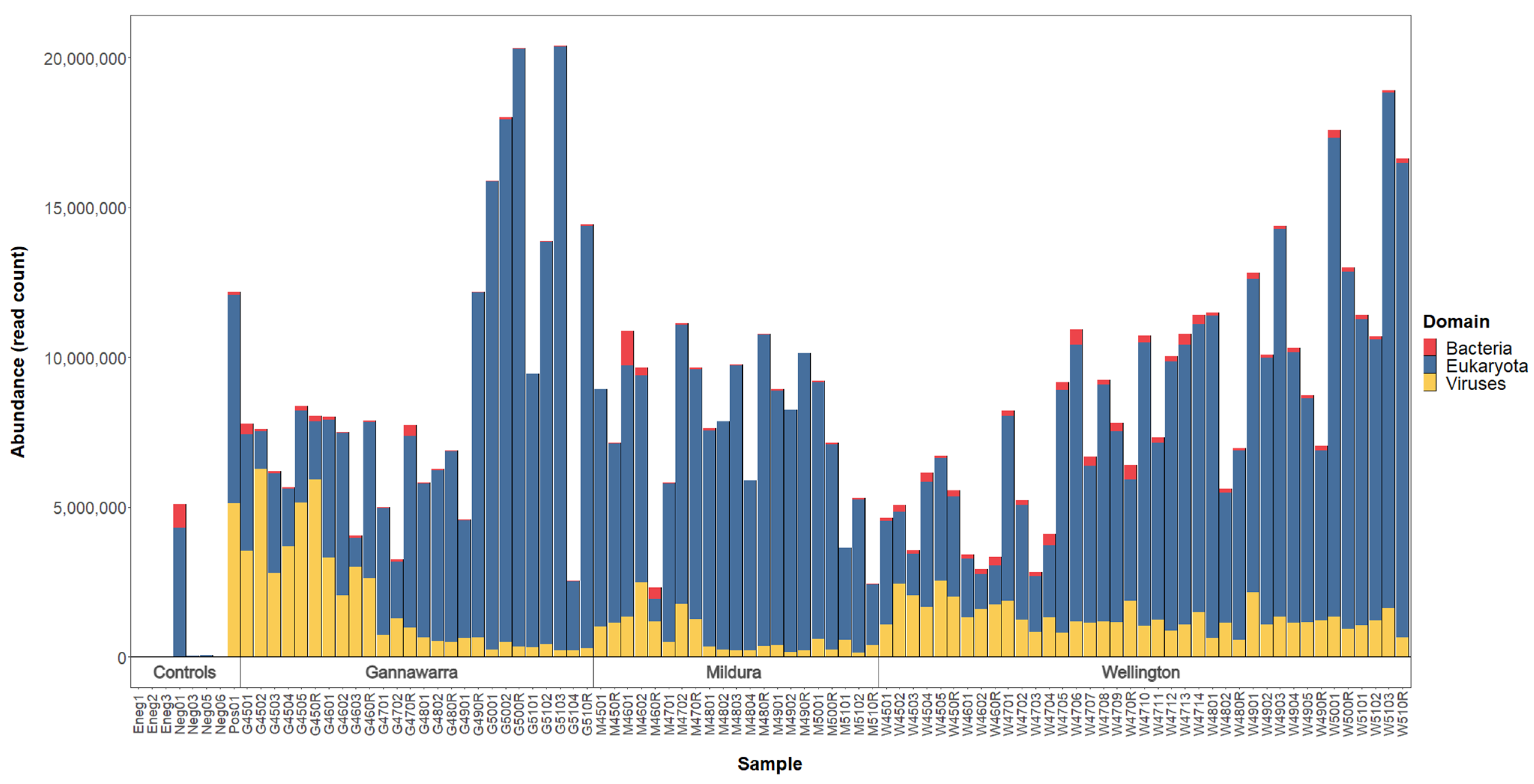
3.1. Sample Preparation and Sequencing
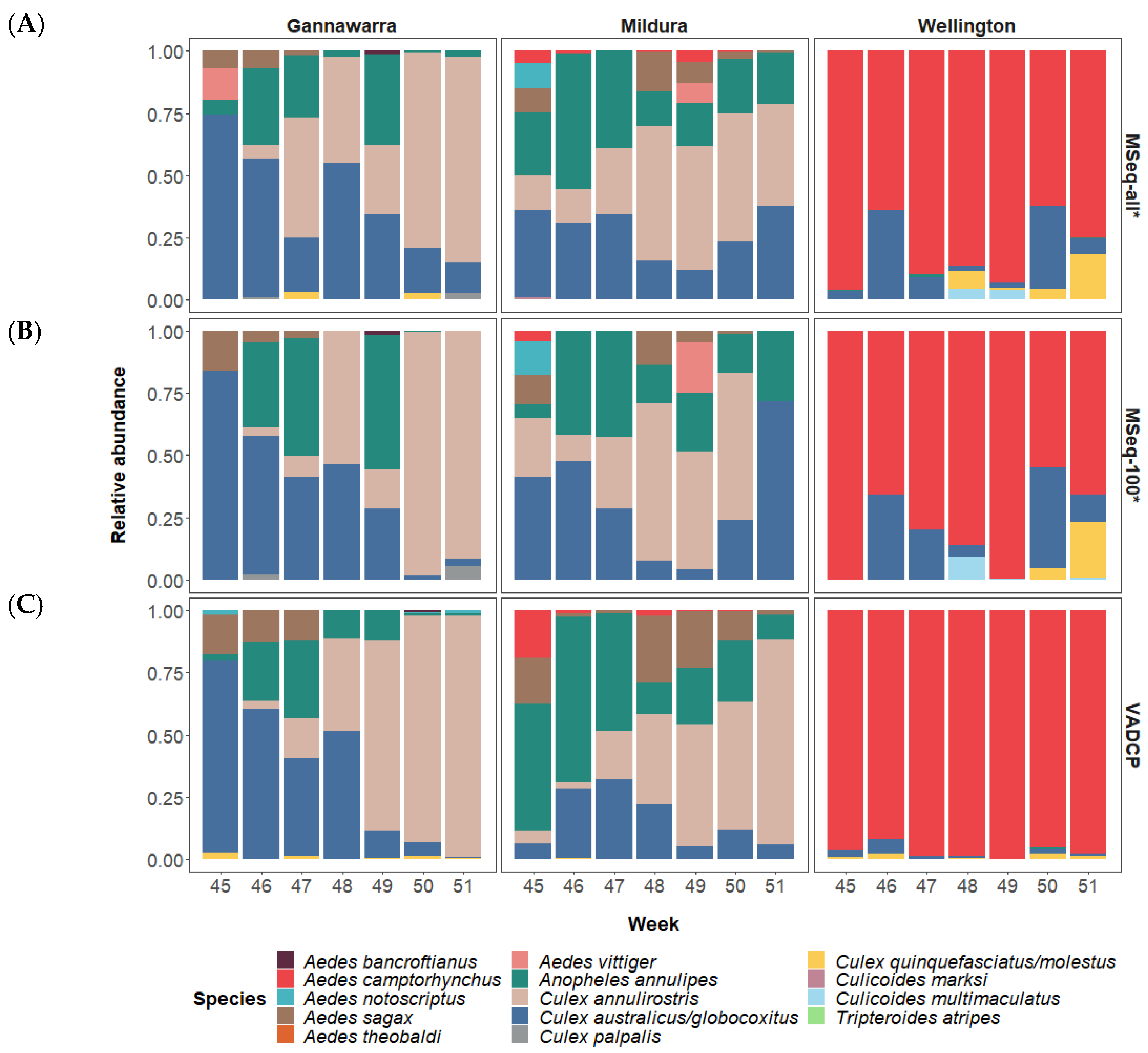
3.2. Mosquito and Biting Midge Species Identification
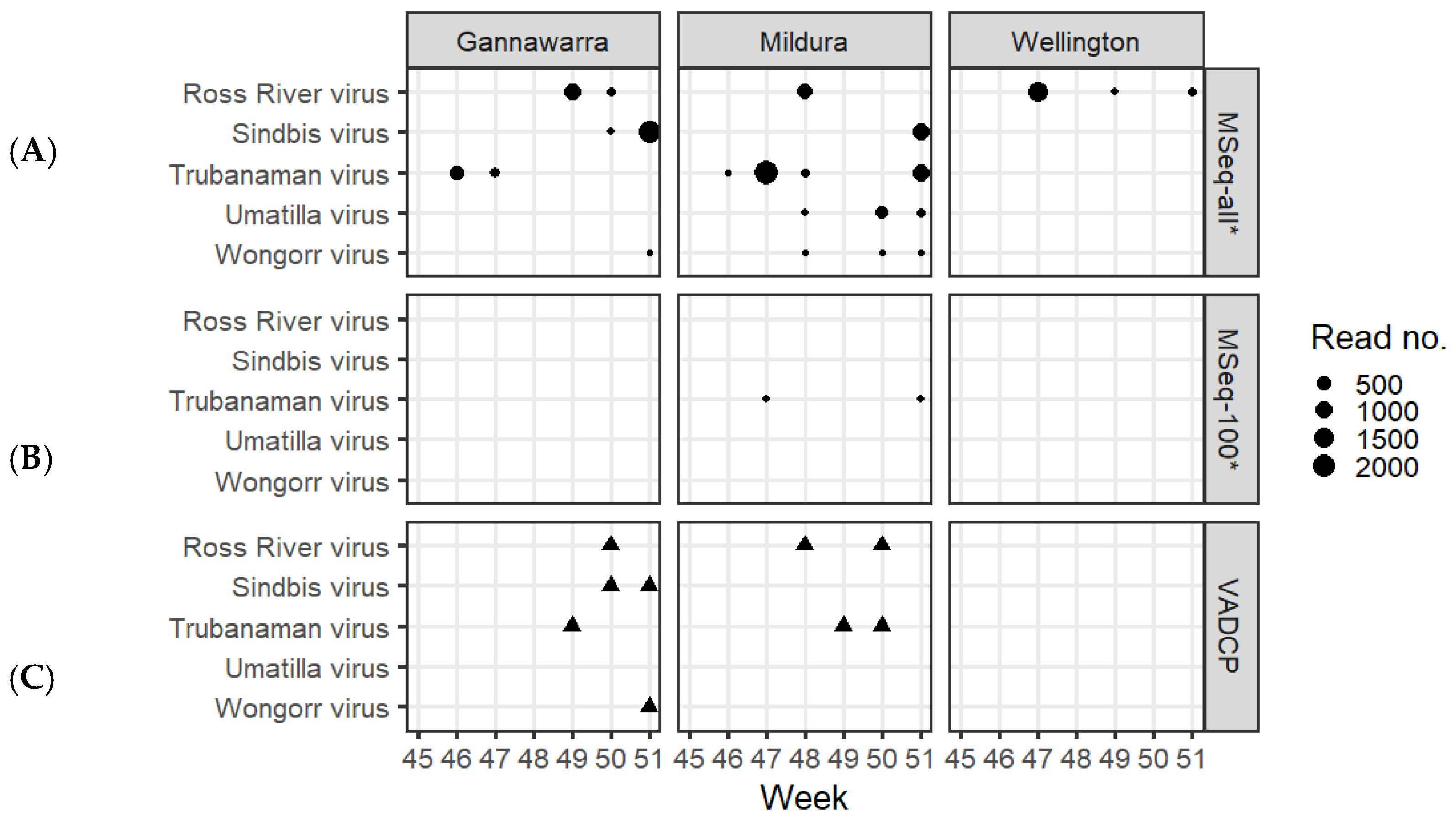
3.3. Assessment of Positive Detection Criteria
3.4. Arbovirus Detection
3.5. Phylogenetic Analysis of Arboviruses
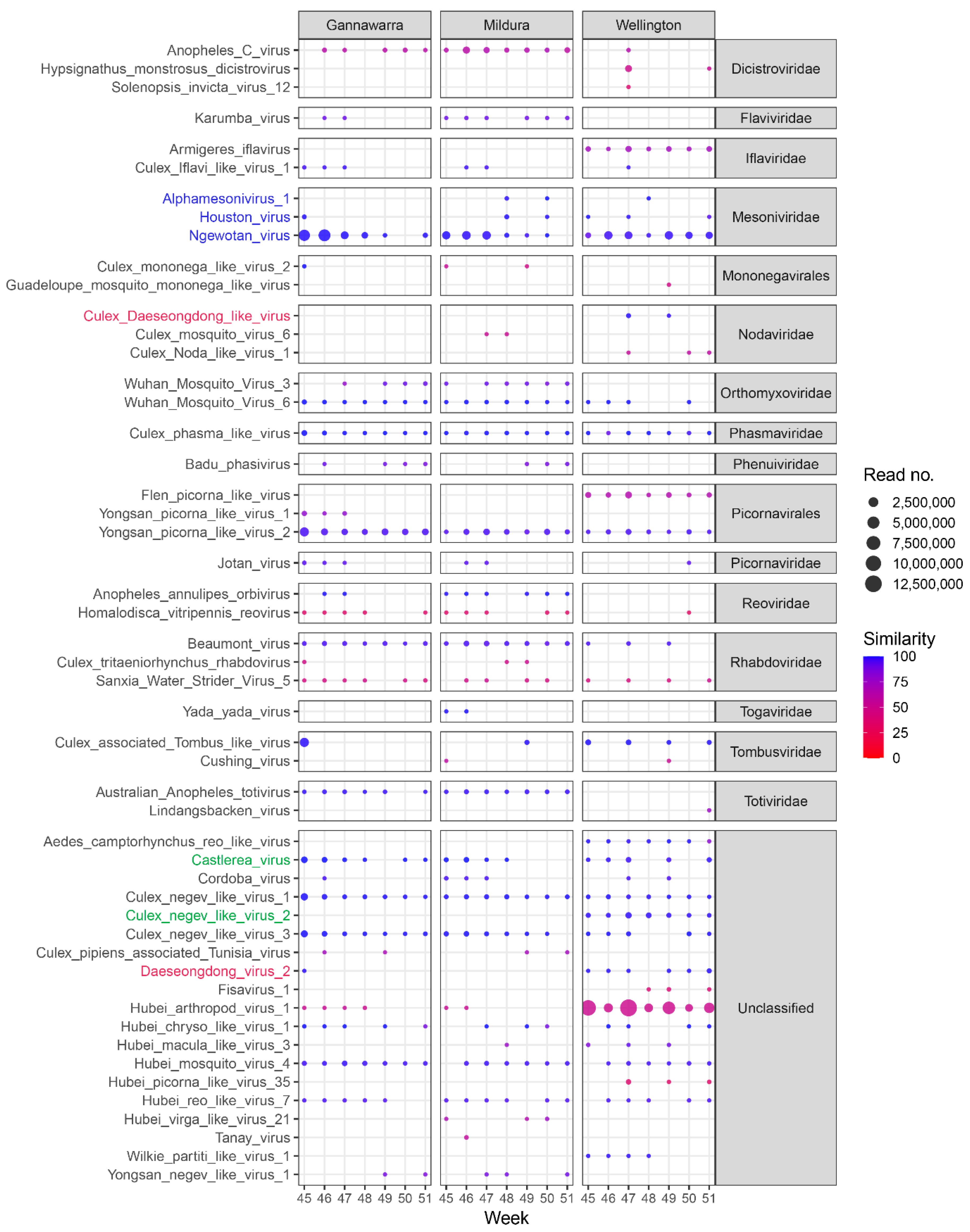
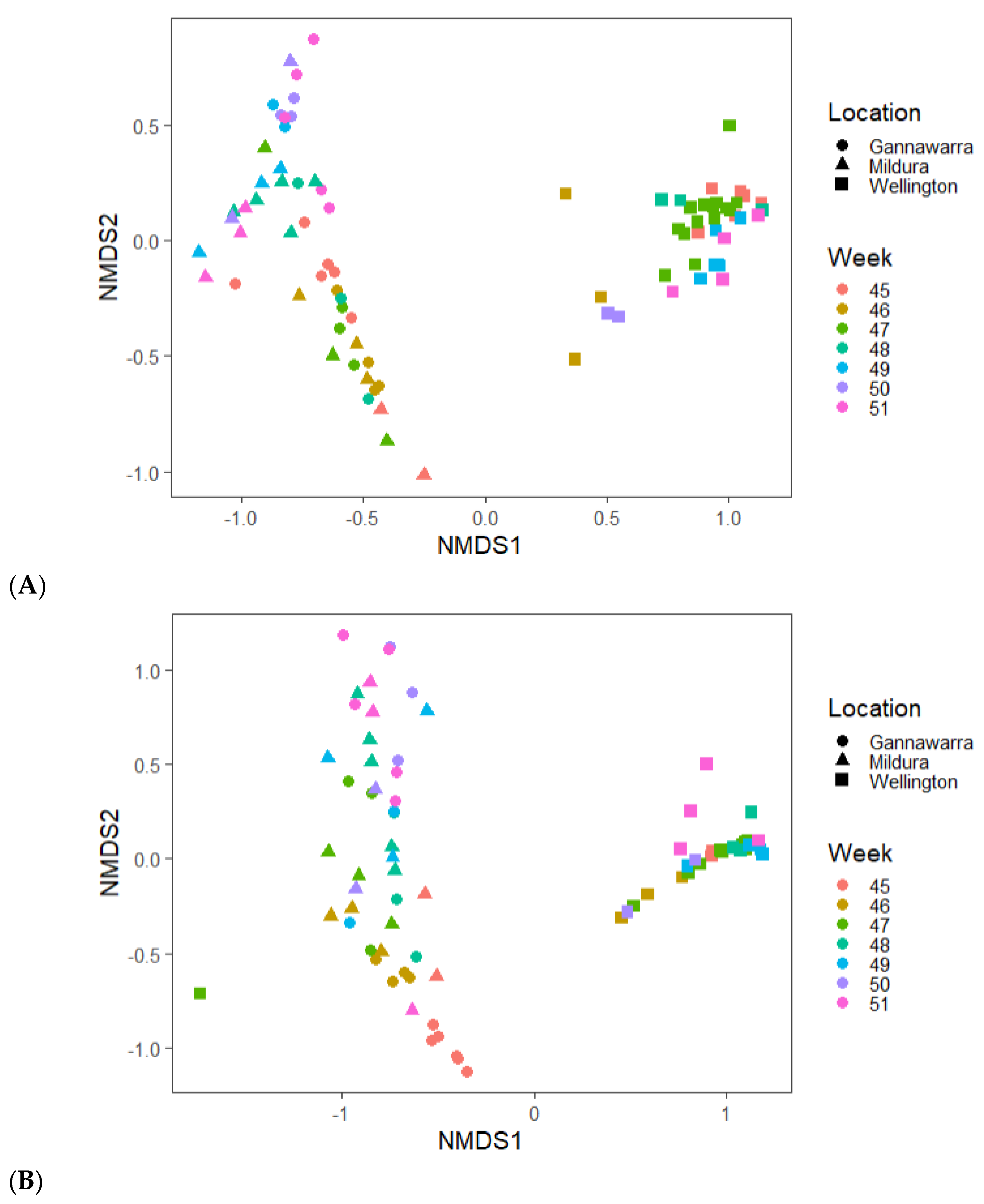
3.6. Virome Ecology
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mayer, S.V.; Tesh, R.B.; Vasilakis, N. The Emergence of Arthropod-Borne Viral Diseases: A Global Prospective on Dengue, Chikungunya and Zika Fevers. Acta Trop. 2017, 166, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Selck, F.W.; Adalja, A.A.; Boddie, C.R. An Estimate of the Global Health Care and Lost Productivity Costs of Dengue. Vector-Borne Zoonotic Dis. 2014, 14, 824–826. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global Trends in Emerging Infectious Diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef] [PubMed]
- Wilder-Smith, A.; Gubler, D.J.; Weaver, S.C.; Monath, T.P.; Heymann, D.L.; Scott, T.W. Epidemic Arboviral Diseases: Priorities for Research and Public Health. Lancet Infect. Dis. 2017, 17, e101–e106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grubaugh, N.D.; Saraf, S.; Gangavarapu, K.; Watts, A.; Tan, A.L.; Oidtman, R.J.; Ladner, J.T.; Oliveira, G.; Matteson, N.L.; Kraemer, M.U.G.; et al. Travel Surveillance and Genomics Uncover a Hidden Zika Outbreak during the Waning Epidemic. Cell 2019, 178, 1057–1071.e11. [Google Scholar] [CrossRef]
- Priyamvada, L.; Quicke, K.M.; Hudson, W.H.; Onlamoon, N.; Sewatanon, J.; Edupuganti, S.; Pattanapanyasat, K.; Chokephaibulkit, K.; Mulligan, M.J.; Wilson, P.C.; et al. Human Antibody Responses after Dengue Virus Infection Are Highly Cross-Reactive to Zika Virus. PNAS 2016, 113, 7852–7857. [Google Scholar] [CrossRef] [Green Version]
- Knope, K.; Doggett, S.L.; Jansen, C.C.; Johansen, C.A.; Kurucz, N.; Feldman, R.; Lynch, S.E.; Hobby, M.P.; Sly, A.; Jardine, A.; et al. Arboviral Diseases and Malaria in Australia, 2014–2015: Annual Report of the National Arbovirus and Malaria Advisory Committee. Commun. Dis. Intell. 2019, 43, 1–69. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Hobson-Peters, J.; Yam, A.W.Y.; Colmant, A.M.G.; McLean, B.J.; Prow, N.A.; Watterson, D.; Hall-Mendelin, S.; Warrilow, D.; Ng, M.-L.; et al. Viral RNA Intermediates as Targets for Detection and Discovery of Novel and Emerging Mosquito-Borne Viruses. PLOS Negl. Trop. Dis. 2015, 9, e0003629. [Google Scholar] [CrossRef] [Green Version]
- Ritchie, S.A.; Pyke, A.T.; Smith, G.A.; Northill, J.A.; Hall, R.A.; van den Hurk, A.F.; Johansen, C.A.; Montgomery, B.L.; Mackenzie, J.S. Field Evaluation of a Sentinel Mosquito (Diptera: Culicidae) Trap System to Detect Japanese Encephalitis in Remote Australia. J. Med. Èntomol. 2003, 40, 249–252. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, G.L.; Nasci, R.S. Detection of West Nile Virus in Large Pools of Mosquitoes. J. Am. Mosq. Control Assoc. 2007, 23, 389–395. [Google Scholar] [CrossRef]
- Gu, W.; Novak, R.J. Short Report: Detection Probability of Arbovirus Infection in Mosquito Populations. Am. J. Trop. Med. Hyg. 2004, 71, 636–638. [Google Scholar] [CrossRef]
- Sozhamannan, S.; Holland, M.Y.; Hall, A.T.; Negrón, D.A.; Ivancich, M.; Koehler, J.W.; Minogue, T.D.; Campbell, C.E.; Berger, W.J.; Christopher, G.W.; et al. Evaluation of Signature Erosion in Ebola Virus Due to Genomic Drift and Its Impact on the Performance of Diagnostic Assays. Viruses 2015, 7, 3130–3154. [Google Scholar] [CrossRef] [Green Version]
- Stellrecht, K.A. The Drift in Molecular Testing for Influenza: Mutations Affecting Assay Performance. J. Clin. Microbiol. 2018, 56, e01531–e17. [Google Scholar] [CrossRef] [Green Version]
- Gaudreault, N.N.; Madden, D.W.; Wilson, W.C.; Trujillo, J.D.; Richt, J.A. African Swine Fever Virus: An Emerging DNA Arbovirus. Front. Vet. Sci. 2020, 7, 215. [Google Scholar] [CrossRef]
- Pollett, S.; Fauver, J.R.; Maljkovic Berry, I.; Melendrez, M.; Morrison, A.; Gillis, L.D.; Johansson, M.A.; Jarman, R.G.; Grubaugh, N.D. Genomic Epidemiology as a Public Health Tool to Combat Mosquito-Borne Virus Outbreaks. J. Infect. Dis. 2020, 221, S308–S318. [Google Scholar] [CrossRef]
- Ramos-Nino, M.E.; Fitzpatrick, D.M.; Eckstrom, K.M.; Tighe, S.; Hattaway, L.M.; Hsueh, A.N.; Stone, D.M.; Dragon, J.A.; Cheetham, S. Metagenomic Analysis of Aedes Aegypti and Culex Quinquefasciatus Mosquitoes from Grenada, West Indies. PLoS ONE 2020, 15, e0231047. [Google Scholar] [CrossRef]
- Chandler, J.A.; Liu, R.M.; Bennett, S.N. RNA Shotgun Metagenomic Sequencing of Northern California (USA) Mosquitoes Uncovers Viruses, Bacteria, and Fungi. Front. Microbiol. 2015, 6, 185. [Google Scholar] [CrossRef] [Green Version]
- Belda, E.; Nanfack-Minkeu, F.; Eiglmeier, K.; Carissimo, G.; Holm, I.; Diallo, M.; Diallo, D.; Vantaux, A.; Kim, S.; Sharakhov, I.V.; et al. De Novo Profiling of RNA Viruses in Anopheles Malaria Vector Mosquitoes from Forest Ecological Zones in Senegal and Cambodia. BMC Genomics 2019, 20, 664. [Google Scholar] [CrossRef] [Green Version]
- Steinegger, M.; Salzberg, S.L. Terminating Contamination: Large-Scale Search Identifies More than 2,000,000 Contaminated Entries in GenBank. Genome Biol. 2020, 21, 115. [Google Scholar] [CrossRef]
- Chiu, C.Y.; Miller, S.A. Clinical Metagenomics. Nat. Rev. Genet. 2019, 20, 341–355. [Google Scholar] [CrossRef]
- Pettersson, J.H.-O.; Shi, M.; Eden, J.-S.; Holmes, E.C.; Hesson, J.C. Meta-Transcriptomic Comparison of the RNA Viromes of the Mosquito Vectors Culex Pipiens and Culex Torrentium in Northern Europe. Viruses 2019, 11, 1033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadeghi, M.; Altan, E.; Deng, X.; Barker, C.M.; Fang, Y.; Coffey, L.L.; Delwart, E. Virome of > 12 Thousand Culex Mosquitoes from throughout California. Virology 2018, 523, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.-S.; Imrie, A.; Holmes, E.C. High-Resolution Metatranscriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. J. Virol. 2017, 91, e00680–e17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakrzewski, M.; Rašić, G.; Darbro, J.; Krause, L.; Poo, Y.S.; Filipović, I.; Parry, R.; Asgari, S.; Devine, G.; Suhrbier, A. Mapping the Virome in Wild-Caught Aedes Aegypti from Cairns and Bangkok. Sci. Rep. 2018, 8, 4690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batovska, J.; Mee, P.T.; Lynch, S.E.; Sawbridge, T.I.; Rodoni, B.C. Sensitivity and Specificity of Metatranscriptomics as an Arbovirus Surveillance Tool. Sci. Rep. 2019, 9, 19398. [Google Scholar] [CrossRef] [Green Version]
- Rohe, D.L.; Fall, R.P. A Miniature Battery Powered CO2 Baited Light Trap for Mosquito-Borne Encephalitis Surveillance. Bull. Soc. Vector Ecol. 1979, 4, 24–27. [Google Scholar]
- Murray-Darling Basin Authority. River Murray Weekly Report for the Week Ending Wednesday, 5 October 2016; Australian Government: Canberra, ACT, Australia, 2016.
- Lynch, S.E.; Mee, P.T.; Batovska, J.; Brown, K.; Crowder, J.; Rodoni, B.C. Mosquito and Arbovirus Biodiversity and Ross River Virus Genomic Epidemiology in Victoria, Australia, 2016–2017. Agriculture Victoria Research, Bundoora, VIC, Australia. 2022; manuscript in preparation. [Google Scholar]
- Illumina Effects of Index Misassignment on Multiplexing and Downstream Analysis. [White paper]. 2017. Available online: https://www.illumina.com/content/dam/illumina-marketing/documents/products/whitepapers/index-hopping-white-paper-770-2017-004.pdf (accessed on 25 November 2019).
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-Length Transcriptome Assembly from RNA-Seq Data without a Reference Genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. ArXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- RStudio Team. RStudio: Integrated Development for R.; RStudio: Boston, MA, USA, 2015. [Google Scholar]
- McMurdie, P.J.; Holmes, S. phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar]
- Mariadassou, M.; Pichon, S.; Ebert, D. Microbial Ecosystems Are Dominated by Specialist Taxa. Ecol. Lett. 2015, 18, 974–982. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Batovska, J.; Blacket, M.J.; Brown, K.; Lynch, S.E. Molecular Identification of Mosquitoes (Diptera: Culicidae) in Southeastern Australia. Ecol. Evol. 2016, 6, 3001–3011. [Google Scholar] [CrossRef] [Green Version]
- Dyce, A.L.; Bellis, G.A.; Muller, M.J. Pictorial Atlas of Australasian Culicoides Wings (Diptera: Ceratopogonidae); Australian Biological Resources Study: Canberra, ACT, Australia, 2007. [Google Scholar]
- Tay, W.T.; Kerr, P.J.; Jermiin, L.S. Population Genetic Structure and Potential Incursion Pathways of the Bluetongue Virus Vector Culicoides Brevitarsis (Diptera: Ceratopogonidae) in Australia. PLoS ONE 2016, 11, e0146699. [Google Scholar] [CrossRef] [Green Version]
- Mackenzie, J.S.; Lindsay, M.D.; Coelen, R.J.; Broom, A.K.; Hall, R.A.; Smith, D.W. Arboviruses Causing Human Disease in the Australasian Zoogeographic Region. Arch. Virol. 1994, 136, 447–467. [Google Scholar] [CrossRef]
- Vasilakis, N.; Tesh, R.B.; Popov, V.L.; Widen, S.G.; Wood, T.G.; Forrester, N.L.; Gonzalez, J.P.; Saluzzo, J.F.; Alkhovsky, S.; Lam, S.K.; et al. Exploiting the Legacy of the Arbovirus Hunters. Viruses 2019, 11, 471. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B. BBMap Short Read Aligner, and Other Bioinformatic Tools. 2017. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 19 November 2019).
- Hall, R.; Prow, N.; Pyke, A. Ross River Virus. In Molecular Detection of Human Viral Pathogens; CRC Press: Boca Raton, FL, USA, 2011; pp. 349–359. [Google Scholar]
- Eshoo, M.W.; Whitehouse, C.A.; Zoll, S.T.; Massire, C.; Pennella, T.-T.D.; Blyn, L.B.; Sampath, R.; Hall, T.A.; Ecker, J.A.; Desai, A.; et al. Direct Broad-Range Detection of Alphaviruses in Mosquito Extracts. Virology 2007, 368, 286–295. [Google Scholar] [CrossRef] [Green Version]
- Cowled, C.; Palacios, G.; Melville, L.; Weir, R.; Walsh, S.; Davis, S.; Gubala, A.; Lipkin, W.I.; Briese, T.; Boyle, D. Genetic and Epidemiological Characterization of Stretch Lagoon Orbivirus, a Novel Orbivirus Isolated from Culex and Aedes Mosquitoes in Northern Australia. J. Gen. Virol. 2009, 90, 1433–1439. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wille, M.; Shi, M.; Klaassen, M.; Hurt, A.C.; Holmes, E.C. Virome Heterogeneity and Connectivity in Waterfowl and Shorebird Communities. ISME J. 2019, 13, 2603–2616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michie, A.; Dhanasekaran, V.; Lindsay, M.D.A.; Neville, P.J.; Nicholson, J.; Jardine, A.; Mackenzie, J.S.; Smith, D.W.; Imrie, A. Genome-Scale Phylogeny and Evolutionary Analysis of Ross River Virus Reveals Periodic Sweeps of Lineage Dominance in Western Australia, 1977-2014. J. Virol. 2020, 94, e01234–e19. [Google Scholar] [CrossRef] [Green Version]
- Pickering, P.; Aaskov, J.G.; Liu, W. Complete Genomic Sequence of an Australian Sindbis Virus Isolated 44 Years Ago Reveals Unique Indels in the E2 and nsP3 Proteins. Microbiol. Resour. Announc. 2019, 8, e00246–e19. [Google Scholar] [CrossRef] [Green Version]
- Saleh, S.M.; Poidinger, M.; Mackenzie, J.S.; Broom, A.K.; Lindsay, M.D.; Hall, R.A. Complete Genomic Sequence of the Australian South-West Genotype of Sindbis Virus: Comparisons with Other Sindbis Strains and Identification of a Unique Deletion in the 3’-Untranslated Region. Virus Genes 2003, 26, 317–327. [Google Scholar] [CrossRef]
- Briese, T.; Williams, D.T.; Kapoor, V.; Diviney, S.M.; Certoma, A.; Wang, J.; Johansen, C.A.; Chowdhary, R.; Mackenzie, J.S.; Lipkin, W.I. Analysis of Arbovirus Isolates from Australia Identifies Novel Bunyaviruses Including a Mapputta Group Virus from Western Australia That Links Gan Gan and Maprik Viruses. PLoS ONE 2016, 11, e0164868. [Google Scholar] [CrossRef] [Green Version]
- Coffey, L.L.; Page, B.L.; Greninger, A.L.; Herring, B.L.; Russell, R.C.; Doggett, S.L.; Haniotis, J.; Wang, C.; Deng, X.; Delwart, E.L. Enhanced Arbovirus Surveillance with Deep Sequencing: Identification of Novel Rhabdoviruses and Bunyaviruses in Australian Mosquitoes. Virology 2014, 448, 146–158. [Google Scholar] [CrossRef]
- Shchetinin, A.M.; Lvov, D.K.; Deriabin, P.G.; Botikov, A.G.; Gitelman, A.K.; Kuhn, J.H.; Alkhovsky, S.V. Genetic and Phylogenetic Characterization of Tataguine and Witwatersrand Viruses and Other Orthobunyaviruses of the Anopheles A, Capim, Guamá, Koongol, Mapputta, Tete, and Turlock Serogroups. Viruses 2015, 7, 5987–6008. [Google Scholar] [CrossRef] [Green Version]
- Ejiri, H.; Kuwata, R.; Tsuda, Y.; Sasaki, T.; Kobayashi, M.; Sato, Y.; Sawabe, K.; Isawa, H. First Isolation and Characterization of a Mosquito-Borne Orbivirus Belonging to the Species Umatilla Virus in East Asia. Arch. Virol. 2014, 159, 2675–2685. [Google Scholar] [CrossRef]
- Bennett, A.J.; Bushmaker, T.; Cameron, K.; Ondzie, A.; Niama, F.R.; Parra, H.-J.; Mombouli, J.-V.; Olson, S.H.; Munster, V.J.; Goldberg, T.L. Diverse RNA Viruses of Arthropod Origin in the Blood of Fruit Bats Suggest a Link Between Bat and Arthropod Viromes. Virology 2019, 528, 64–72. [Google Scholar] [CrossRef]
- Reuter, G.; Pankovics, P.; Delwart, E.; Boros, Á. A Novel Posavirus-Related Single-Stranded RNA Virus from Fish (Cyprinus Carpio). Arch. Virol. 2015, 160, 565–568. [Google Scholar] [CrossRef]
- Colmant, A.M.G.; O’Brien, C.A.; Newton, N.D.; Watterson, D.; Hardy, J.; Coulibaly, F.; Bielefeldt-Ohmann, H.; Warrilow, D.; Huang, B.; Paramitha, D.; et al. Novel Monoclonal Antibodies against Australian Strains of Negeviruses and Insights into Virus Structure, Replication and Host-Restriction. J. Gen. Virol. 2020, 101, 440–452. [Google Scholar] [CrossRef]
- Jansen, C.C.; Hemmerter, S.; van den Hurk, A.F.; Whelan, P.I.; Beebe, N.W. Morphological versus Molecular Identification of Culex Annulirostris Skuse and Culex Palpalis Taylor: Key Members of the Culex Sitiens (Diptera: Culicidae) Subgroup in Australasia. Aust. J. Entomol. 2013, 52, 356–362. [Google Scholar] [CrossRef]
- Ekrem, T.; Willassen, E.; Stur, E. A Comprehensive DNA Sequence Library is Essential for Identification with DNA Barcodes. Mol. Phylogenetics Evol. 2007, 43, 530–542. [Google Scholar] [CrossRef]
- Harley, D.; Sleigh, A.; Ritchie, S. Ross River Virus Transmission, Infection, and Disease: A Cross-Disciplinary Review. Clin. Microbiol. Rev. 2001, 14, 909–932. [Google Scholar] [CrossRef] [Green Version]
- Boughton, C.R.; Hawkes, R.A.; Naim, H.M. Arbovirus Infection in Humans in NSW: Seroprevalence and Pathogenicity of Certain Australian Bunyaviruses. Aust. New Zealand J. Med. 1990, 20, 51–55. [Google Scholar] [CrossRef]
- Doherty, R.L.; Whitehead, R.H.; Wetters, E.J.; Gorman, B.M.; Carley, J.G. A Survey of Antibody to 10 Arboviruses (Koongol Group, Mapputta Group and Ungrouped) Isolated in Queensland. Trans. R. Soc. Trop. Med. Hyg. 1970, 64, 748–753. [Google Scholar] [CrossRef]
- Belaganahalli, M.N.; Maan, S.; Maan, N.S.; Tesh, R.; Attoui, H.; Mertens, P.P.C. Umatilla Virus Genome Sequencing and Phylogenetic Analysis: Identification of Stretch Lagoon Orbivirus as a New Member of the Umatilla Virus Species. PLoS ONE 2011, 6, e23605. [Google Scholar] [CrossRef] [Green Version]
- Doherty, R.L.; Carley, J.G.; Standfast, H.A.; Dyce, A.L.; Kay, B.H.; Snowdon, W.A. Isolation of Arboviruses from Mosquitoes, Biting Midges, Sandflies and Vertebrates Collected in Queensland, 1969 and 1970. Trans. R. Soc. Trop. Med. Hyg. 1973, 67, 536–543. [Google Scholar] [CrossRef] [PubMed]
- Coffey, L.L.; Forrester, N.; Tsetsarkin, K.; Vasilakis, N.; Weaver, S.C. Factors Shaping the Adaptive Landscape for Arboviruses: Implications for the Emergence of Disease. Future Microbiol. 2013, 8, 155–176. [Google Scholar] [CrossRef] [PubMed]
- Tangudu, C.S.; Charles, J.; Hurt, S.L.; Dunphy, B.M.; Smith, R.C.; Bartholomay, L.C.; Blitvich, B.J. Skunk River Virus, a Novel Orbivirus Isolated from Aedes Trivittatus in the United States. J. Gen. Virol. 2019, 100, 295–300. [Google Scholar] [CrossRef] [PubMed]
- Pyke, A.T.; Warrilow, D. Archival Collections Are Important in the Study of the Biology, Diversity, and Evolution of Arboviruses. Evol. Bioinform. Online 2016, 12, EBO–S40569. [Google Scholar] [CrossRef] [Green Version]
- Lemmon, G.H.; Gardner, S.N. Predicting the Sensitivity and Specificity of Published Real-Time PCR Assays. Ann. Clin. Microbiol. Antimicrob. 2008, 7, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrader, C.; Schielke, A.; Ellerbroek, L.; Johne, R. PCR Inhibitors–Occurrence, Properties and Removal. J. Appl. Microbiol. 2012, 113, 1014–1026. [Google Scholar] [CrossRef]
- Bibby, K.; Peccia, J. Identification of Viral Pathogen Diversity in Sewage Sludge by Metagenome Analysis. Environ. Sci. Technol. 2013, 47, 1945–1951. [Google Scholar] [CrossRef] [Green Version]
- Fernandez-Cassi, X.; Timoneda, N.; Gonzales-Gustavson, E.; Abril, J.F.; Bofill-Mas, S.; Girones, R. A Metagenomic Assessment of Viral Contamination on Fresh Parsley Plants Irrigated with Fecally Tainted River Water. Int. J. Food Microbiol. 2017, 257, 80–90. [Google Scholar] [CrossRef]
- Fernandez-Cassi, X.; Timoneda, N.; Martínez-Puchol, S.; Rusiñol, M.; Rodriguez-Manzano, J.; Figuerola, N.; Bofill-Mas, S.; Abril, J.F.; Girones, R. Metagenomics for the Study of Viruses in Urban Sewage as a Tool for Public Health Surveillance. Sci. Total Environ. 2018, 618, 870–880. [Google Scholar] [CrossRef]
- Schlaberg, R.; Chiu, C.Y.; Miller, S.; Procop, G.W.; Weinstock, G.; Professional Practice Committee and Committee on Laboratory Practices of the American Society for Microbiology; Microbiology Resource Committee of the College of American Pathologists. Validation of Metagenomic Next-Generation Sequencing Tests for Universal Pathogen Detection. Arch. Pathol. Lab. Med. 2017, 141, 776–786. [Google Scholar] [CrossRef] [Green Version]
- Piper, A.M.; Batovska, J.; Cogan, N.O.I.; Weiss, J.; Cunningham, J.P.; Rodoni, B.C.; Blacket, M.J. Prospects and Challenges of Implementing DNA Metabarcoding for High-Throughput Insect Surveillance. GigaScience 2019, 8, giz092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kramer, L.D.; Chiles, R.E.; Do, T.D.; Fallah, H.M. Detection of St. Louis Encephalitis and Western Equine Encephalomyelitis RNA in Mosquitoes Tested without Maintenance of a Cold Chain. J. Am. Mosq. Control. Assoc. 2001, 17, 213–215. [Google Scholar] [PubMed]
- Mavale, M.; Sudeep, A.; Gokhale, M.; Hundekar, S.; Parashar, D.; Ghodke, Y.; Arankalle, V.; Mishra, A.C. Persistence of Viral RNA in Chikungunya Virus-Infected Aedes Aegypti (Diptera: Culicidae) Mosquitoes after Prolonged Storage at 28 °C. Am. J. Trop. Med. Hyg. 2012, 86, 178–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, A.J.M.; Stanley, G.; Sinha, R.; Weissman, I.L.; Sandberg, R. Computational Correction of Index Switching in Multiplexed Sequencing Libraries. Nat. Methods 2018, 15, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented Genomic Diversity of RNA Viruses in Arthropods Reveals the Ancestry of Negative-Sense RNA Viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Hobson-Peters, J.; Harrison, J.J.; Watterson, D.; Hazlewood, J.E.; Vet, L.J.; Newton, N.D.; Warrilow, D.; Colmant, A.M.G.; Taylor, C.; Huang, B.; et al. A Recombinant Platform for Flavivirus Vaccines and Diagnostics Using Chimeras of a New Insect-Specific Virus. Sci. Transl. Med. 2019, 11, eaax7888. [Google Scholar] [CrossRef]
- Öhlund, P.; Lundén, H.; Blomström, A.-L. Insect-Specific Virus Evolution and Potential Effects on Vector Competence. Virus Genes 2019, 55, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Batson, J.; Dudas, G.; Haas-Stapleton, E.; Kistler, A.L.; Li, L.M.; Logan, P.; Ratnasiri, K.; Retallack, H. Single Mosquito Metatranscriptomics Recovers Mosquito Species, Blood Meal Sources, and Microbial Cargo, Including Viral Dark Matter. BioRxiv 2020, 2020-02. [Google Scholar] [CrossRef]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef]
- Simmonds, P. Methods for Virus Classification and the Challenge of Incorporating Metagenomic Sequence Data. J. Gen. Virol. 2015, 96, 1193–1206. [Google Scholar] [CrossRef]
- Dacheux, L.; Cervantes-Gonzalez, M.; Guigon, G.; Thiberge, J.-M.; Vandenbogaert, M.; Maufrais, C.; Caro, V.; Bourhy, H. A Preliminary Study of Viral Metagenomics of French Bat Species in Contact with Humans: Identification of New Mammalian Viruses. PLoS ONE 2014, 9, e87194. [Google Scholar] [CrossRef]
- Harvey, E.; Rose, K.; Eden, J.-S.; Lo, N.; Abeyasuriya, T.; Shi, M.; Doggett, S.L.; Holmes, E.C. Extensive Diversity of RNA Viruses in Australian Ticks. J. Virol. 2019, 93, e01358–e18. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Category | Recommendation | Outcomes |
|---|---|---|
| Sampling | Sequence all the mosquitoes collected from surveillance traps. | Increased likelihood of detecting arboviral activity. |
| Investigate other sample types for metatranscriptomic sequencing (e.g., gravid mosquitoes, FTA cards) | Less sequencing of uninfected mosquitoes, thereby improving arbovirus detection sensitivity. | |
| Laboratory protocol | Develop an in-house ribosomal RNA depletion protocol. | Greater ability to customise depletion, thereby improving arbovirus detection sensitivity; decreased assay cost. |
| Use unique dual indexing to multiplex samples. | Reduced index cross-talk, thereby improving arbovirus detection sensitivity. | |
| Automate library preparation where possible. | Increased multiplexing capacity; decreased assay cost and turnaround time. | |
| Use ultra-high-throughput sequencing (e.g., Illumina NovaSeq). | Increased multiplexing capacity; decreased assay cost; faster turnaround than HiSeq. | |
| Bioinformatics | Establish a user-friendly bioinformatics pipeline. | Less reliance on specialised bioinformatics expertise. |
| Develop a tool to assess the risk of novel or unclassified viruses. | Faster, less complex data analysis with relevant reporting for public health. | |
| Formulate an organised and cost-effective storage plan for high volumes of sequencing data. | Ability to repurpose or re-analyse past metatranscriptomic surveillance data with updated databases and bioinformatics. | |
| Reference databases | Establish a DNA barcode database of local mosquito species. | Comprehensive identification of mosquito species in surveillance traps. |
| Acquire whole genome sequences of arbovirus isolates for inclusion in databases used for screening data. | Improved arbovirus detection sensitivity; high-resolution phylogenetics to determine arbovirus origins and dispersal. | |
| Curate a contamination database by sequencing samples from laboratory surfaces and reagents. | Improved ability to distinguish real signal from background or contamination. | |
| Quality control | Include negative and positive controls. | Detection of contamination; ability to assess assay validity. |
| Standardise laboratory and bioinformatics protocols. | Consistent, reproducible surveillance results of known sensitivity and specificity. | |
| Regularly validate assay sensitivity and specificity in response to protocol modifications. | Enables protocol updates while ensuring adequate assay sensitivity and appropriate detection thresholds. | |
| Confirm important arbovirus detections with RT-qPCR. | Confidence in reliability of arbovirus detections for public health reporting. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batovska, J.; Mee, P.T.; Sawbridge, T.I.; Rodoni, B.C.; Lynch, S.E. Enhanced Arbovirus Surveillance with High-Throughput Metatranscriptomic Processing of Field-Collected Mosquitoes. Viruses 2022, 14, 2759. https://doi.org/10.3390/v14122759
Batovska J, Mee PT, Sawbridge TI, Rodoni BC, Lynch SE. Enhanced Arbovirus Surveillance with High-Throughput Metatranscriptomic Processing of Field-Collected Mosquitoes. Viruses. 2022; 14(12):2759. https://doi.org/10.3390/v14122759
Chicago/Turabian StyleBatovska, Jana, Peter T. Mee, Tim I. Sawbridge, Brendan C. Rodoni, and Stacey E. Lynch. 2022. "Enhanced Arbovirus Surveillance with High-Throughput Metatranscriptomic Processing of Field-Collected Mosquitoes" Viruses 14, no. 12: 2759. https://doi.org/10.3390/v14122759
APA StyleBatovska, J., Mee, P. T., Sawbridge, T. I., Rodoni, B. C., & Lynch, S. E. (2022). Enhanced Arbovirus Surveillance with High-Throughput Metatranscriptomic Processing of Field-Collected Mosquitoes. Viruses, 14(12), 2759. https://doi.org/10.3390/v14122759