
The Discovery of a New Mimivirus Isolate in Association with Virophage-Transpoviron Elements in Brazil Highlights the Main Genomic and Evolutionary Features of This Tripartite System

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viral Isolation, Multiplication, and Purification
2.2. Transmission Electron Microscopy
2.3. Sequencing, Assembly, and Annotation
2.4. Phylogeny Analysis
2.5. DNA Extraction and PCR
3. Results and Discussion
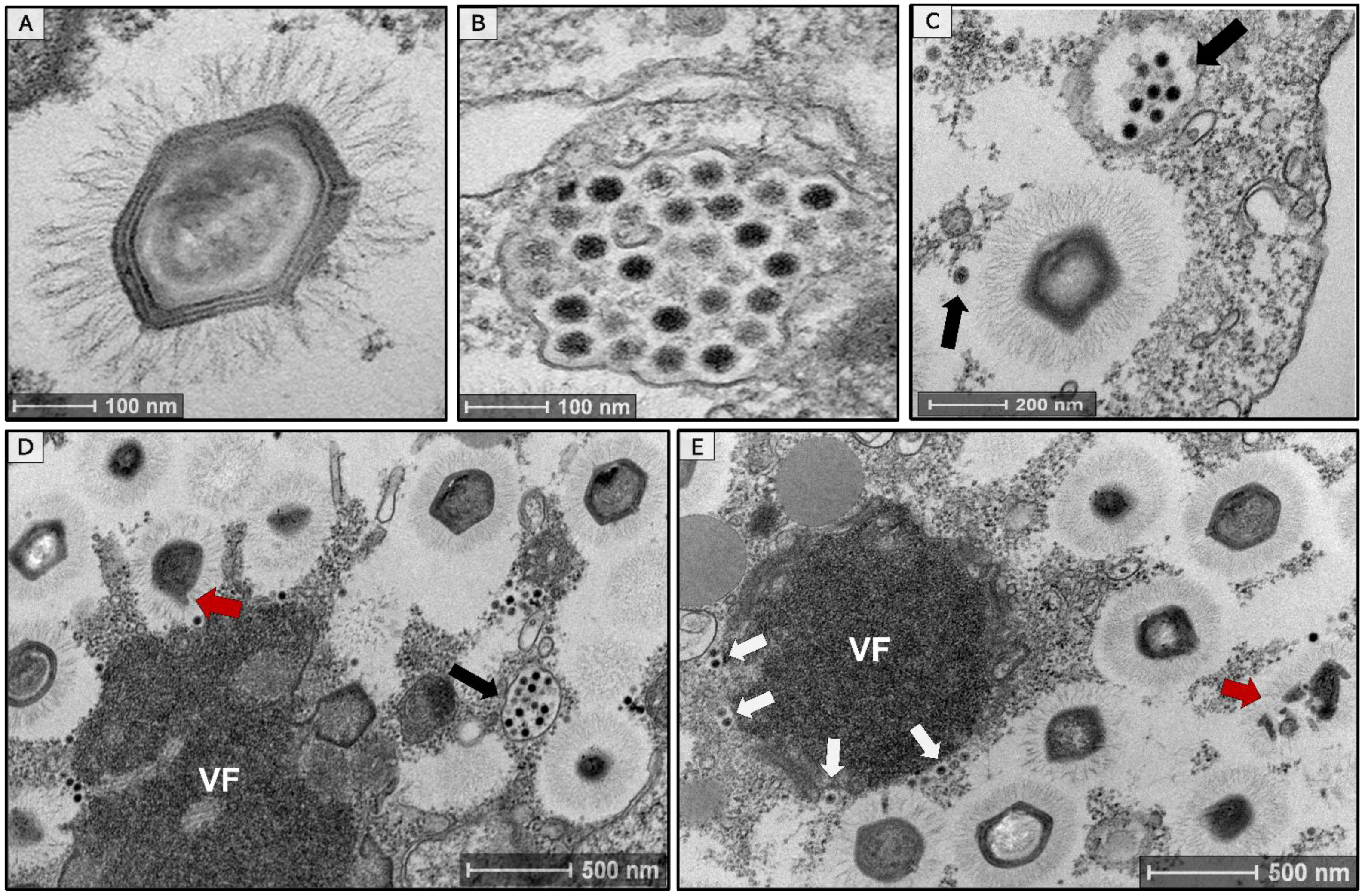
3.1. Isolation of a Tripartite Mimivirus-Virophage-Transpoviron System in Brazil
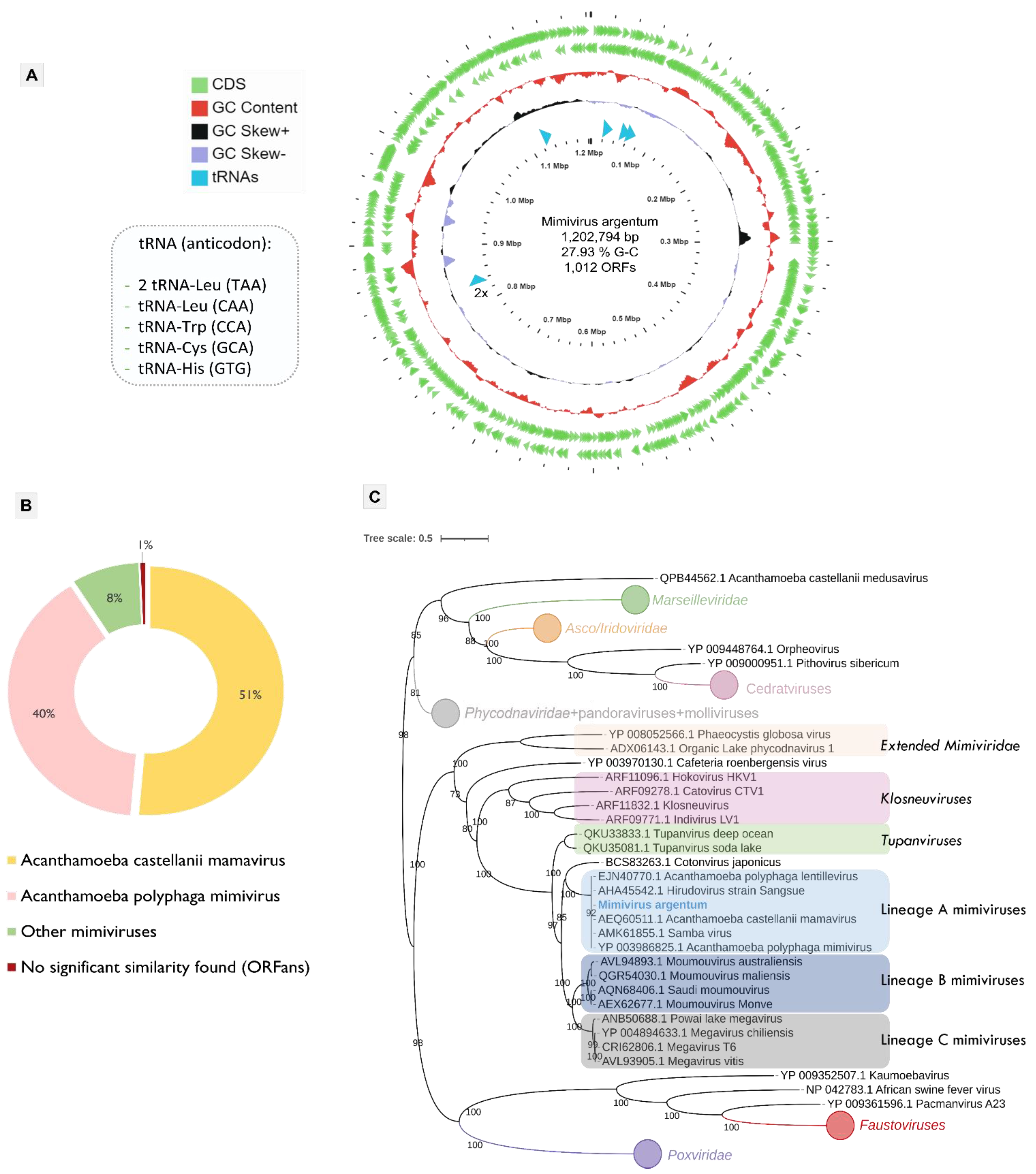
3.2. Mimivirus Argentum Genome
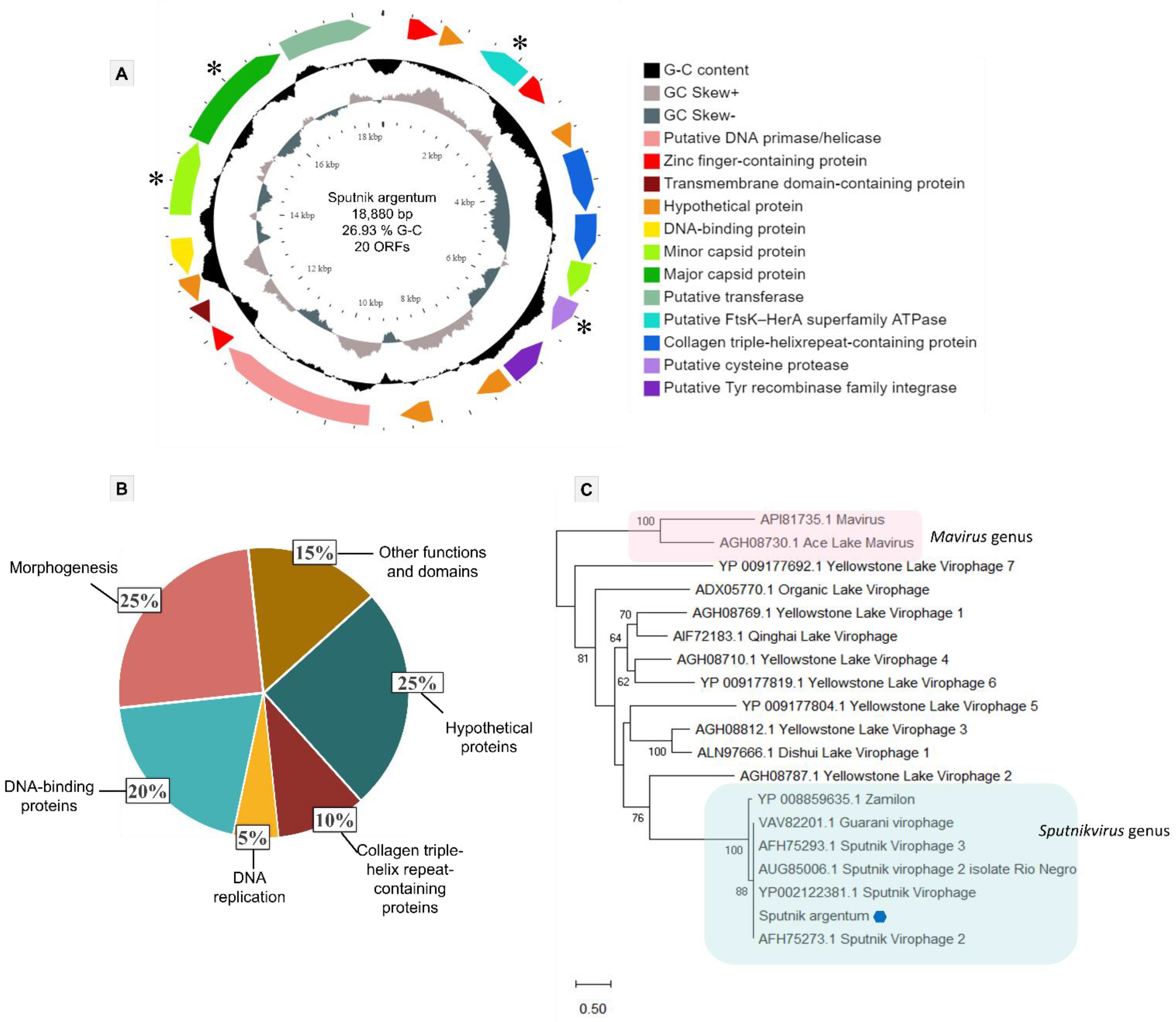
3.3. Sputnik Argentum Genome
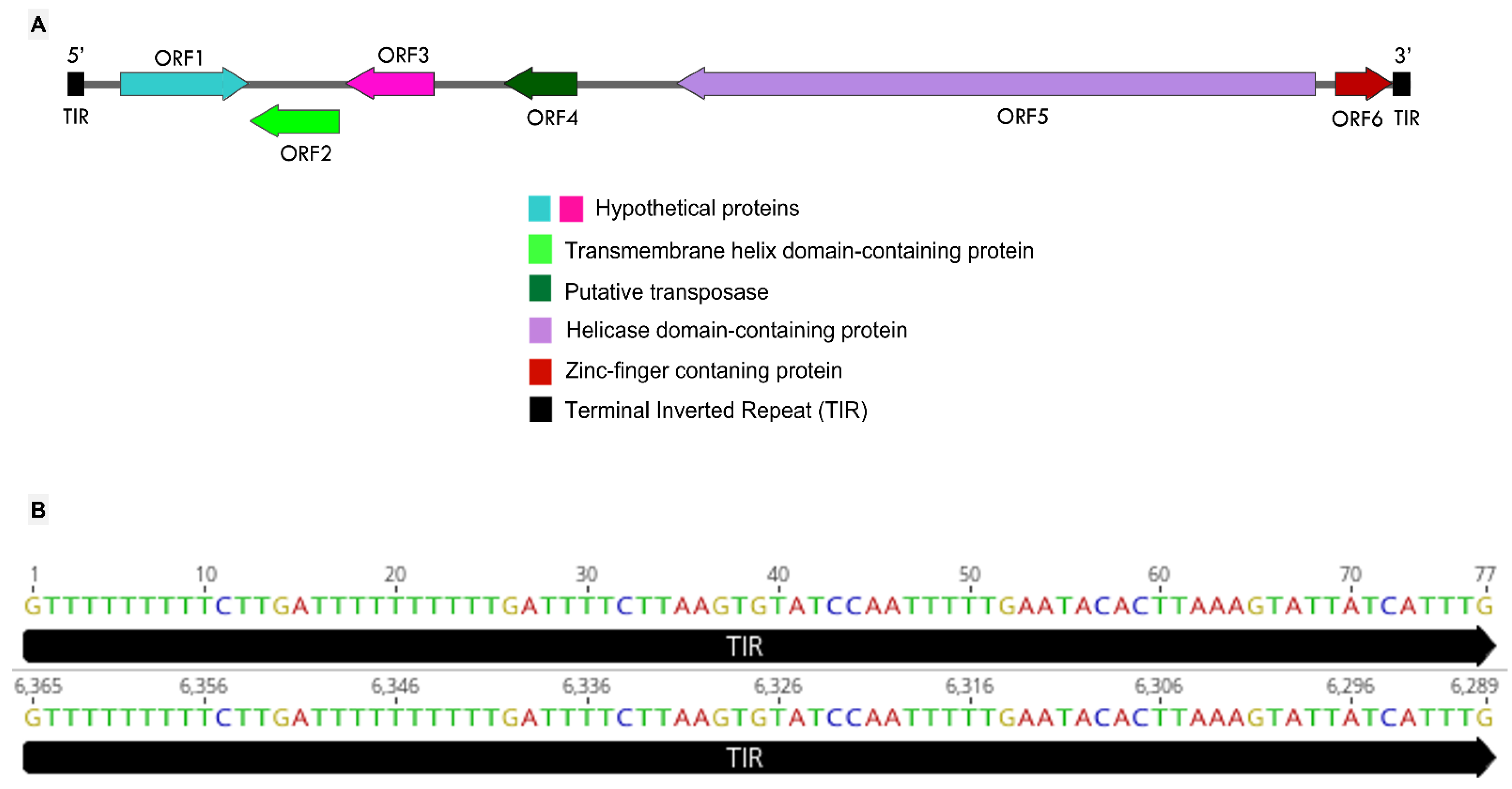
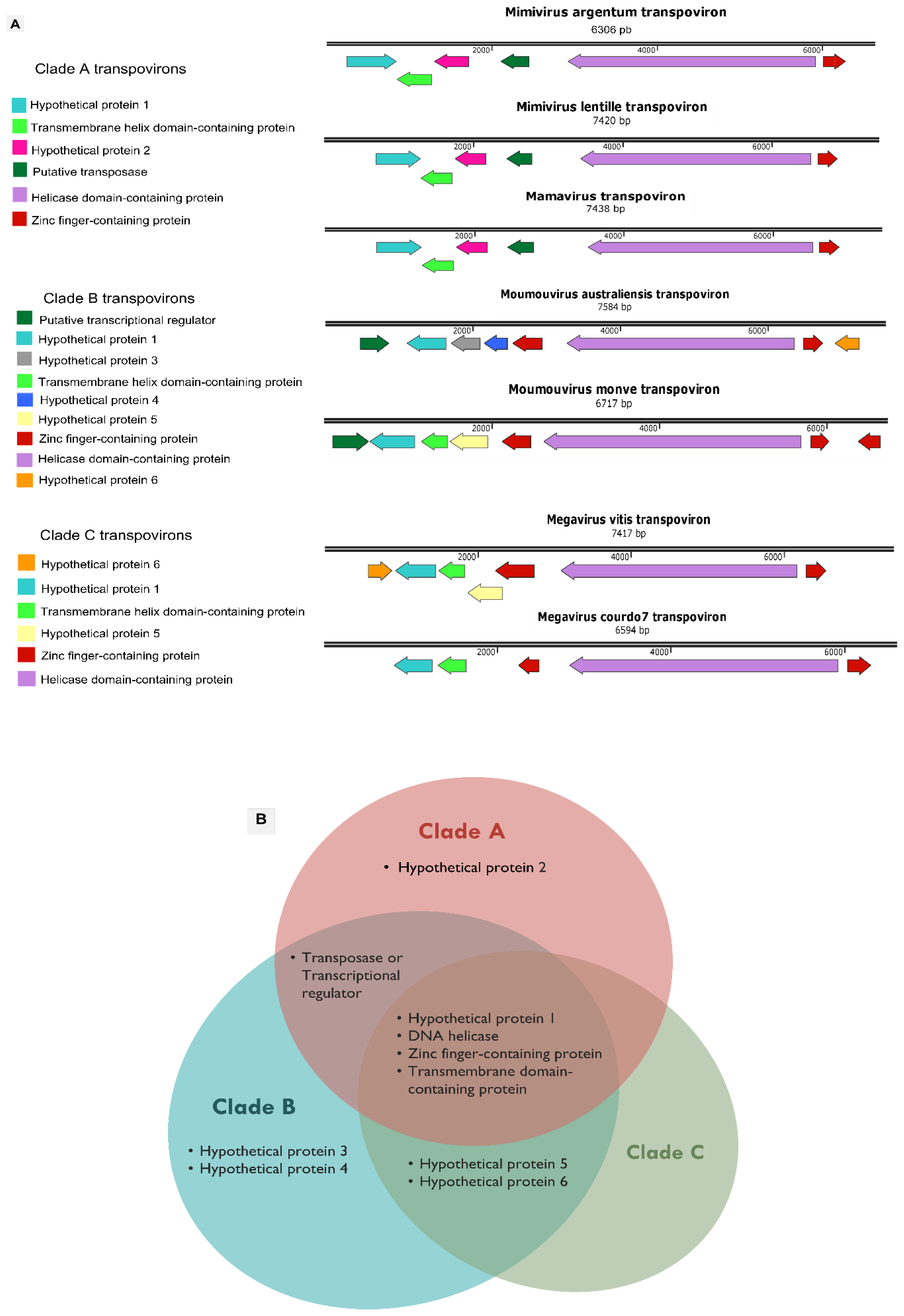
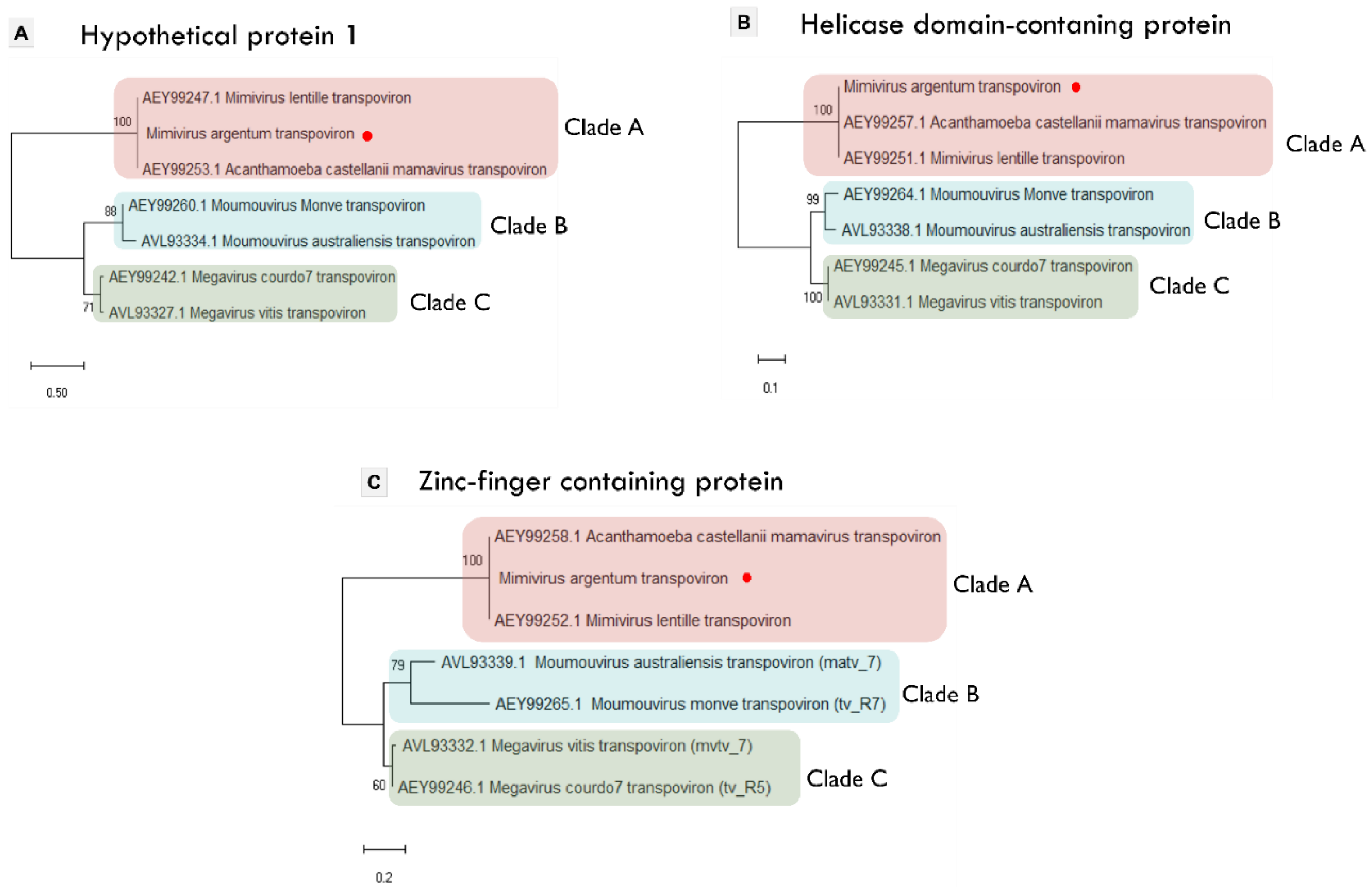
3.4. Mimivirus Argentum-Associated Transpoviron
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lwoff, A. The Concept of Virus. J. Gen. Microbiol. 1957, 17, 239–253. [Google Scholar] [CrossRef] [Green Version]
- La Scola, B.; Audic, S.; Robert, C.; Jungang, L.; de Lamballerie, X.; Drancourt, M.; Birtles, R.; Claverie, J.-M.; Raoult, D. A Giant Virus in Amoebae. Science 2003, 299, 2033. [Google Scholar] [CrossRef]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; La Scola, B.; Suzan, M.; Claverie, J.-M. The 1.2-Megabase Genome Sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef]
- Colson, P.; Yutin, N.; Shabalina, S.A.; Robert, C.; Fournous, G.; La Scola, B.; Raoult, D.; Koonin, E.V. Viruses with More than 1000 Genes: Mamavirus, a New Acanthamoeba Polyphaga Mimivirus Strain, and Reannotation of Mimivirus Genes. Genome Biol. Evol. 2011, 3, 737–742. [Google Scholar] [CrossRef]
- Colson, P.; de Lamballerie, X.; Fournous, G.; Raoult, D. Reclassification of Giant Viruses Composing a Fourth Domain of Life in the New Order Megavirales. Intervirology 2012, 55, 321–332. [Google Scholar] [CrossRef]
- Yoosuf, N.; Yutin, N.; Colson, P.; Shabalina, S.A.; Pagnier, I.; Robert, C.; Azza, S.; Klose, T.; Wong, J.; Rossmann, M.G.; et al. Related Giant Viruses in Distant Locations and Different Habitats: Acanthamoeba Polyphaga Moumouvirus Represents a Third Lineage of the Mimiviridae That Is Close to the Megavirus Lineage. Genome Biol. Evol. 2012, 4, 1324–1330. [Google Scholar] [CrossRef] [Green Version]
- Arslan, D.; Legendre, M.; Seltzer, V.; Abergel, C.; Claverie, J.-M. Distant Mimivirus Relative with a Larger Genome Highlights the Fundamental Features of Megaviridae. Proc. Natl. Acad. Sci. USA 2011, 108, 17486–17491. [Google Scholar] [CrossRef] [Green Version]
- Abrahão, J.; Silva, L.; Silva, L.S.; Khalil, J.Y.B.; Rodrigues, R.; Arantes, T.; Assis, F.; Boratto, P.; Andrade, M.; Kroon, E.G.; et al. Tailed Giant Tupanvirus Possesses the Most Complete Translational Apparatus of the Known Virosphere. Nat. Commun. 2018, 9, 749. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, R.A.L.; Mougari, S.; Colson, P.; La Scola, B.; Abrahão, J.S. “Tupanvirus”, a New Genus in the Family Mimiviridae. Arch. Virol. 2019, 164, 325–331. [Google Scholar] [CrossRef] [Green Version]
- Schulz, F.; Yutin, N.; Ivanova, N.N.; Ortega, D.R.; Lee, T.K.; Vierheilig, J.; Daims, H.; Horn, M.; Wagner, M.; Jensen, G.J.; et al. Giant Viruses with an Expanded Complement of Translation System Components. Science 2017, 356, 82–85. [Google Scholar] [CrossRef] [Green Version]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. 2020, 84, e00061-19. [Google Scholar] [CrossRef]
- Iyer, L.M.; Aravind, L.; Koonin, E.V. Common Origin of Four Diverse Families of Large Eukaryotic DNA Viruses. J. Virol. 2001, 75, 11720–11734. [Google Scholar] [CrossRef] [Green Version]
- Boyer, M.; Yutin, N.; Pagnier, I.; Barrassi, L.; Fournous, G.; Espinosa, L.; Robert, C.; Azza, S.; Sun, S.; Rossmann, M.G.; et al. Giant Marseillevirus Highlights the Role of Amoebae as a Melting Pot in Emergence of Chimeric Microorganisms. Proc. Natl. Acad. Sci. USA 2009, 106, 21848–21853. [Google Scholar] [CrossRef] [Green Version]
- Philippe, N.; Legendre, M.; Doutre, G.; Couté, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba Viruses with Genomes up to 2.5 Mb Reaching That of Parasitic Eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef] [Green Version]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-Thousand-Year-Old Distant Relative of Giant Icosahedral DNA Viruses with a Pandoravirus Morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 4274–4279. [Google Scholar] [CrossRef] [Green Version]
- La Scola, B.; Desnues, C.; Pagnier, I.; Robert, C.; Barrassi, L.; Fournous, G.; Merchat, M.; Suzan-Monti, M.; Forterre, P.; Koonin, E.; et al. The Virophage as a Unique Parasite of the Giant Mimivirus. Nature 2008, 455, 100–104. [Google Scholar] [CrossRef]
- Duponchel, S.; Fischer, M.G. Viva Lavidaviruses! Five Features of Virophages That Parasitize Giant DNA Viruses. PLoS Pathog. 2019, 15, e1007592. [Google Scholar] [CrossRef]
- Desnues, C.; La Scola, B.; Yutin, N.; Fournous, G.; Robert, C.; Azza, S.; Jardot, P.; Monteil, S.; Campocasso, A.; Koonin, E.V.; et al. Provirophages and Transpovirons as the Diverse Mobilome of Giant Viruses. Proc. Natl. Acad. Sci. USA 2012, 109, 18078–18083. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.G.; Hackl, T. Host Genome Integration and Giant Virus-Induced Reactivation of the Virophage Mavirus. Nature 2016, 540, 288–291. [Google Scholar] [CrossRef]
- Krupovic, M.; Kuhn, J.H.; Fischer, M.G. A Classification System for Virophages and Satellite Viruses. Arch. Virol. 2016, 161, 233–247. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.G.; Suttle, C.A. A Virophage at the Origin of Large DNA Transposons. Science 2011, 332, 231–234. [Google Scholar] [CrossRef]
- Gaia, M.; Benamar, S.; Boughalmi, M.; Pagnier, I.; Croce, O.; Colson, P.; Raoult, D.; La Scola, B. Zamilon, a Novel Virophage with Mimiviridae Host Specificity. PLoS ONE 2014, 9, e94923. [Google Scholar] [CrossRef]
- Zhou, J.; Zhang, W.; Yan, S.; Xiao, J.; Zhang, Y.; Li, B.; Pan, Y.; Wang, Y. Diversity of Virophages in Metagenomic Data Sets. J. Virol. 2013, 87, 4225–4236. [Google Scholar] [CrossRef] [Green Version]
- Paez-Espino, D.; Zhou, J.; Roux, S.; Nayfach, S.; Pavlopoulos, G.A.; Schulz, F.; McMahon, K.D.; Walsh, D.; Woyke, T.; Ivanova, N.N.; et al. Diversity, Evolution, and Classification of Virophages Uncovered through Global Metagenomics. Microbiome 2019, 7, 157. [Google Scholar] [CrossRef] [Green Version]
- Yutin, N.; Raoult, D.; Koonin, E.V. Virophages, Polintons, and Transpovirons: A Complex Evolutionary Network of Diverse Selfish Genetic Elements with Different Reproduction Strategies. Virol. J. 2013, 10, 158. [Google Scholar] [CrossRef] [Green Version]
- Siefert, J.L. Defining the Mobilome. Methods Mol. Biol. 2009, 532, 13–27. [Google Scholar] [CrossRef]
- Filée, J. Giant Viruses and Their Mobile Genetic Elements: The Molecular Symbiosis Hypothesis. Curr. Opin. Virol. 2018, 33, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Jeudy, S.; Bertaux, L.; Alempic, J.-M.; Lartigue, A.; Legendre, M.; Belmudes, L.; Santini, S.; Philippe, N.; Beucher, L.; Biondi, E.G.; et al. Exploration of the Propagation of Transpovirons within Mimiviridae Reveals a Unique Example of Commensalism in the Viral World. ISME J. 2020, 14, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Andrade, A.C.D.S.P.; Arantes, T.S.; Rodrigues, R.A.L.; Machado, T.B.; Dornas, F.P.; Landell, M.F.; Furst, C.; Borges, L.G.A.; Dutra, L.A.L.; Almeida, G.; et al. Ubiquitous Giants: A Plethora of Giant Viruses Found in Brazil and Antarctica. Virol. J. 2018, 15, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, L.J.; Muench, H. A Simple Method of Estimating Fifty per Cent Endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinform. Oxf. Engl. 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. J. Comput. Mol. Cell Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef]
- Bosi, E.; Donati, B.; Galardini, M.; Brunetti, S.; Sagot, M.-F.; Lió, P.; Crescenzi, P.; Fani, R.; Fondi, M. MeDuSa: A Multi-Draft Based Scaffolder. Bioinform. Oxf. Engl. 2015, 31, 2443–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besemer, J.; Borodovsky, M. GeneMark: Web Software for Gene Finding in Prokaryotes, Eukaryotes and Viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef] [Green Version]
- Laslett, D.; Canback, B. ARAGORN, a Program to Detect TRNA Genes and TmRNA Genes in Nucleotide Sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred Interactive Server for Protein Homology Detection and Structure Prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [Green Version]
- Warburton, P.E.; Giordano, J.; Cheung, F.; Gelfand, Y.; Benson, G. Inverted Repeat Structure of the Human Genome: The X-Chromosome Contains a Preponderance of Large, Highly Homologous Inverted Repeats That Contain Testes Genes. Genome Res. 2004, 14, 1861–1869. [Google Scholar] [CrossRef] [Green Version]
- Grant, J.R.; Stothard, P. The CGView Server: A Comparative Genomics Tool for Circular Genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple Sequence Alignment with High Accuracy and High Throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letunic, I.; Bork, P. Interactive Tree Of Life (ITOL): An Online Tool for Phylogenetic Tree Display and Annotation. Bioinform. Oxf. Engl. 2007, 23, 127–128. [Google Scholar] [CrossRef] [Green Version]
- Desnues, C.; Raoult, D. Inside the Lifestyle of the Virophage. Intervirology 2010, 53, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Andrade, A.C.D.S.P.; Rodrigues, R.A.L.; Oliveira, G.P.; Andrade, K.R.; Bonjardim, C.A.; La Scola, B.; Kroon, E.G.; Abrahão, J.S. Filling Knowledge Gaps for Mimivirus Entry, Uncoating, and Morphogenesis. J. Virol. 2017, 91, e01335-17. [Google Scholar] [CrossRef] [Green Version]
- Dornas, F.P.; Khalil, J.Y.B.; Pagnier, I.; Raoult, D.; Abrahão, J.; La Scola, B. Isolation of New Brazilian Giant Viruses from Environmental Samples Using a Panel of Protozoa. Front. Microbiol. 2015, 6, 1086. [Google Scholar] [CrossRef] [Green Version]
- Fischer, M.G. The Virophage Family Lavidaviridae. Curr. Issues Mol. Biol. 2021, 40, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Wicker, T.; Sabot, F.; Hua-Van, A.; Bennetzen, J.L.; Capy, P.; Chalhoub, B.; Flavell, A.; Leroy, P.; Morgante, M.; Panaud, O.; et al. A Unified Classification System for Eukaryotic Transposable Elements. Nat. Rev. Genet. 2007, 8, 973–982. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Forward | Reverse | Expected Amplicon Sizes |
|---|---|---|---|
| Initial integration region (IIR) | 5′ TATCACCCTTAGTACCCTTG 3′ | 5′ GCAGTGACAAAATACCCATT 3′ | 778 bp |
| Final integration region (FIR) | 5′ CCACAATTAGGGCATTCAC 3′ | 5′ GGAAGCGAAGGTATTAAAGG 3′ | 889 bp |
| Virophage’s ORF 12 | 5′ GCATACTGAAGAGAGTGCCG 3′ | 5′ AGGAAAAGAAAGAGGAACACCAG 3′ | 574 bp |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azevedo, B.L.d.; Júnior, J.P.A.; Ullmann, L.S.; Rodrigues, R.A.L.; Abrahão, J.S. The Discovery of a New Mimivirus Isolate in Association with Virophage-Transpoviron Elements in Brazil Highlights the Main Genomic and Evolutionary Features of This Tripartite System. Viruses 2022, 14, 206. https://doi.org/10.3390/v14020206
Azevedo BLd, Júnior JPA, Ullmann LS, Rodrigues RAL, Abrahão JS. The Discovery of a New Mimivirus Isolate in Association with Virophage-Transpoviron Elements in Brazil Highlights the Main Genomic and Evolutionary Features of This Tripartite System. Viruses. 2022; 14(2):206. https://doi.org/10.3390/v14020206
Chicago/Turabian StyleAzevedo, Bruna Luiza de, João Pessoa Araújo Júnior, Leila Sabrina Ullmann, Rodrigo Araújo Lima Rodrigues, and Jônatas Santos Abrahão. 2022. "The Discovery of a New Mimivirus Isolate in Association with Virophage-Transpoviron Elements in Brazil Highlights the Main Genomic and Evolutionary Features of This Tripartite System" Viruses 14, no. 2: 206. https://doi.org/10.3390/v14020206
APA StyleAzevedo, B. L. d., Júnior, J. P. A., Ullmann, L. S., Rodrigues, R. A. L., & Abrahão, J. S. (2022). The Discovery of a New Mimivirus Isolate in Association with Virophage-Transpoviron Elements in Brazil Highlights the Main Genomic and Evolutionary Features of This Tripartite System. Viruses, 14(2), 206. https://doi.org/10.3390/v14020206