
The Analogs of Furanyl Methylidene Rhodanine Exhibit Broad-Spectrum Inhibitory and Inactivating Activities against Enveloped Viruses, including SARS-CoV-2 and Its Variants

 , ,
, ,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
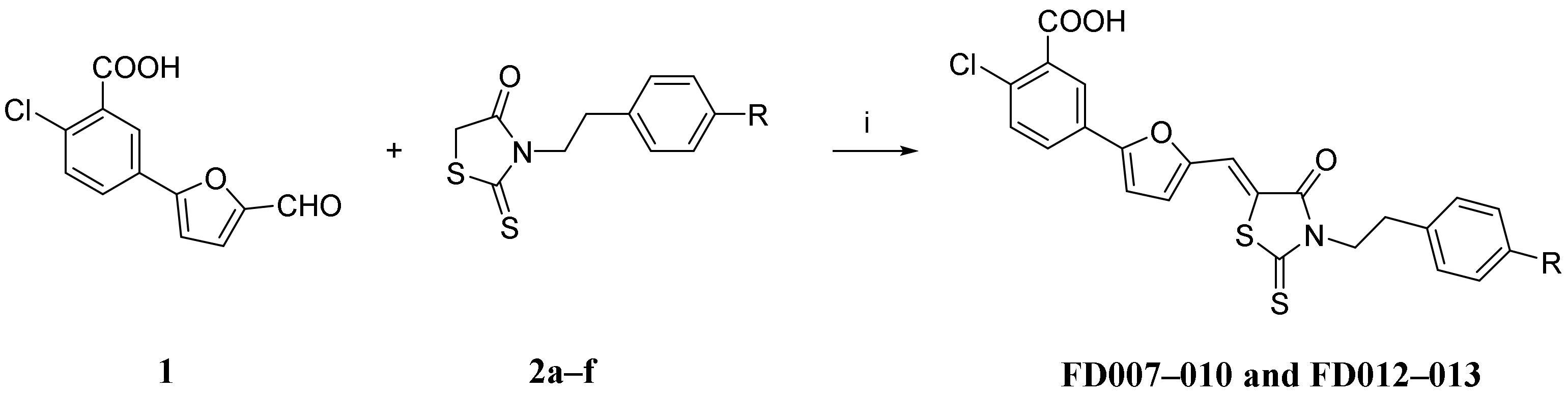
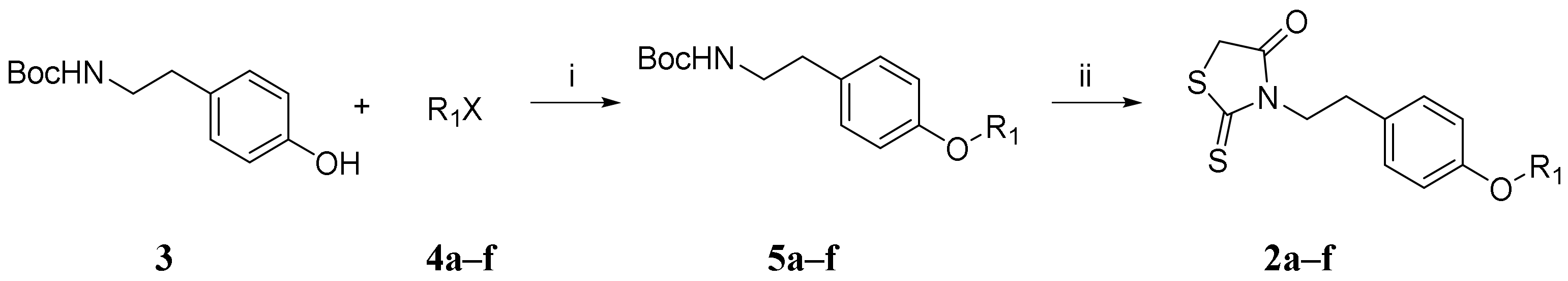
2.1. Synthesis and Characterization of Novel Furanyl Methylidene Rhodanine Analogs FD007–010 and FD012–013
2.2. Cells and Virus
2.3. Inhibition of Pseudovirus (PsV) Infection
2.4. Cytotoxicity
2.5. Inhibition of Authentic Virus Infection
2.6. Inactivation of Cell-Free Virions
2.7. Inhibition of HIV-1 6-HB Formation
2.8. Sucrose Density Gradient Assay to Evaluate Viral Inactivation
3. Results
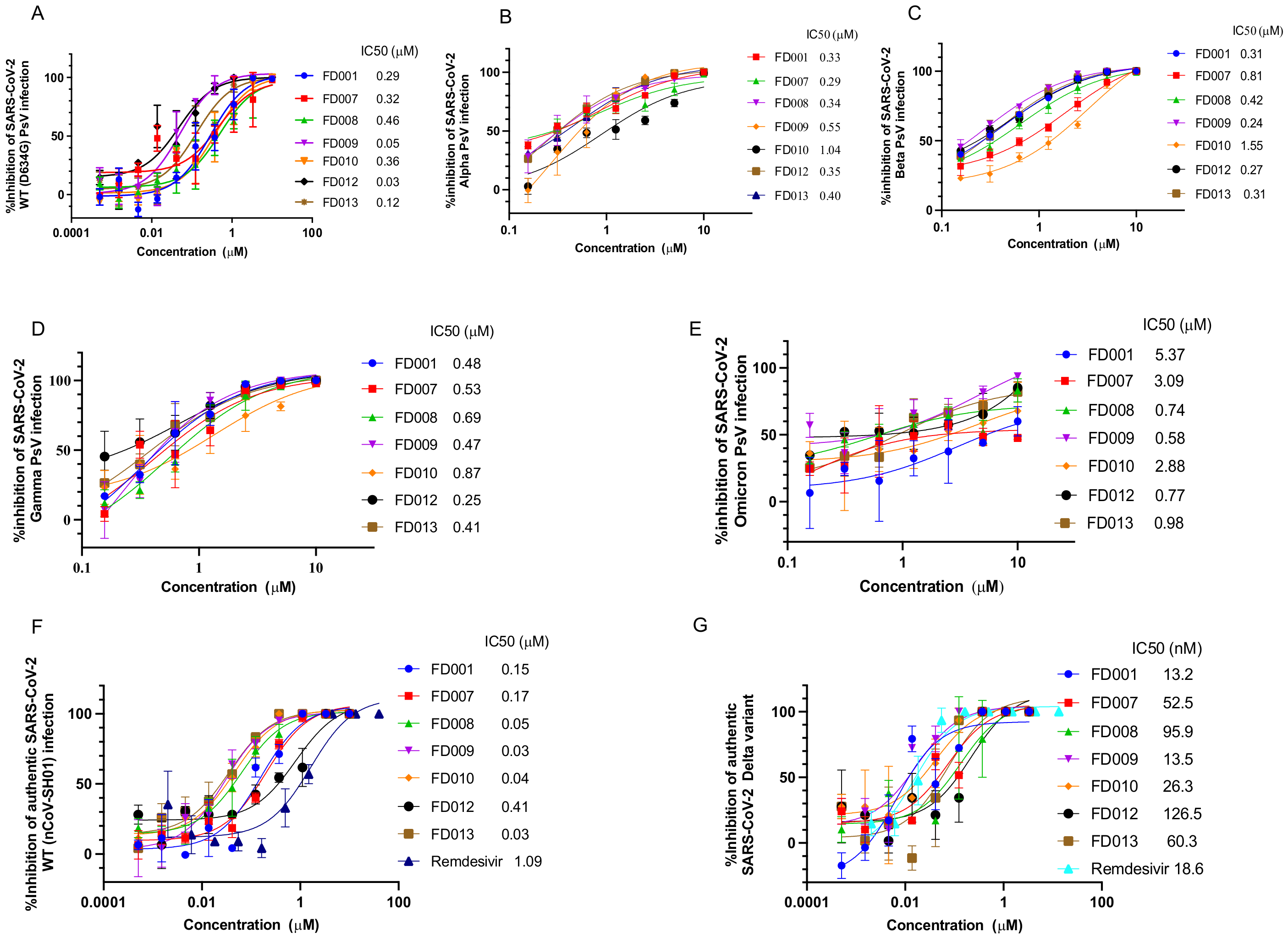
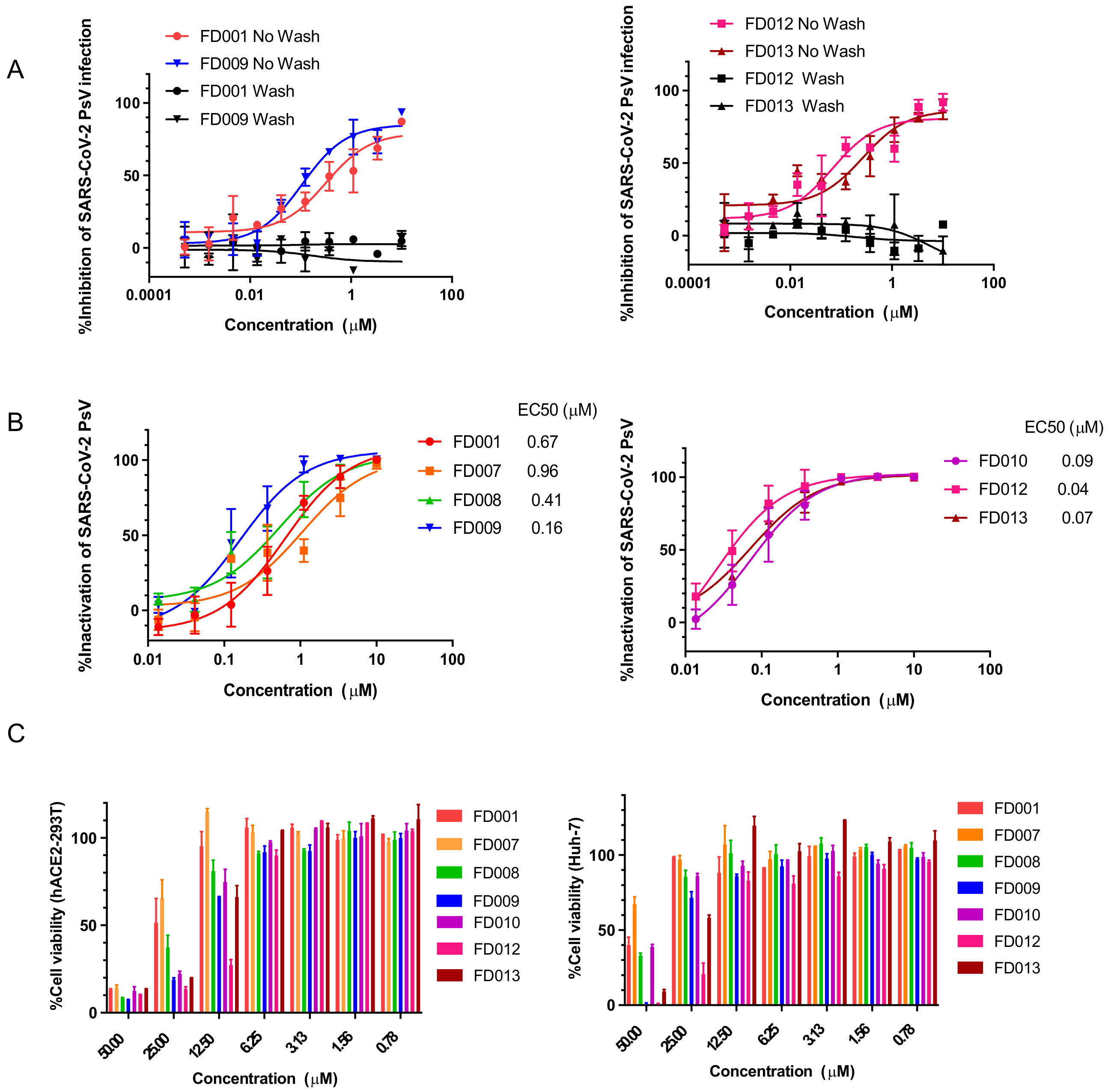
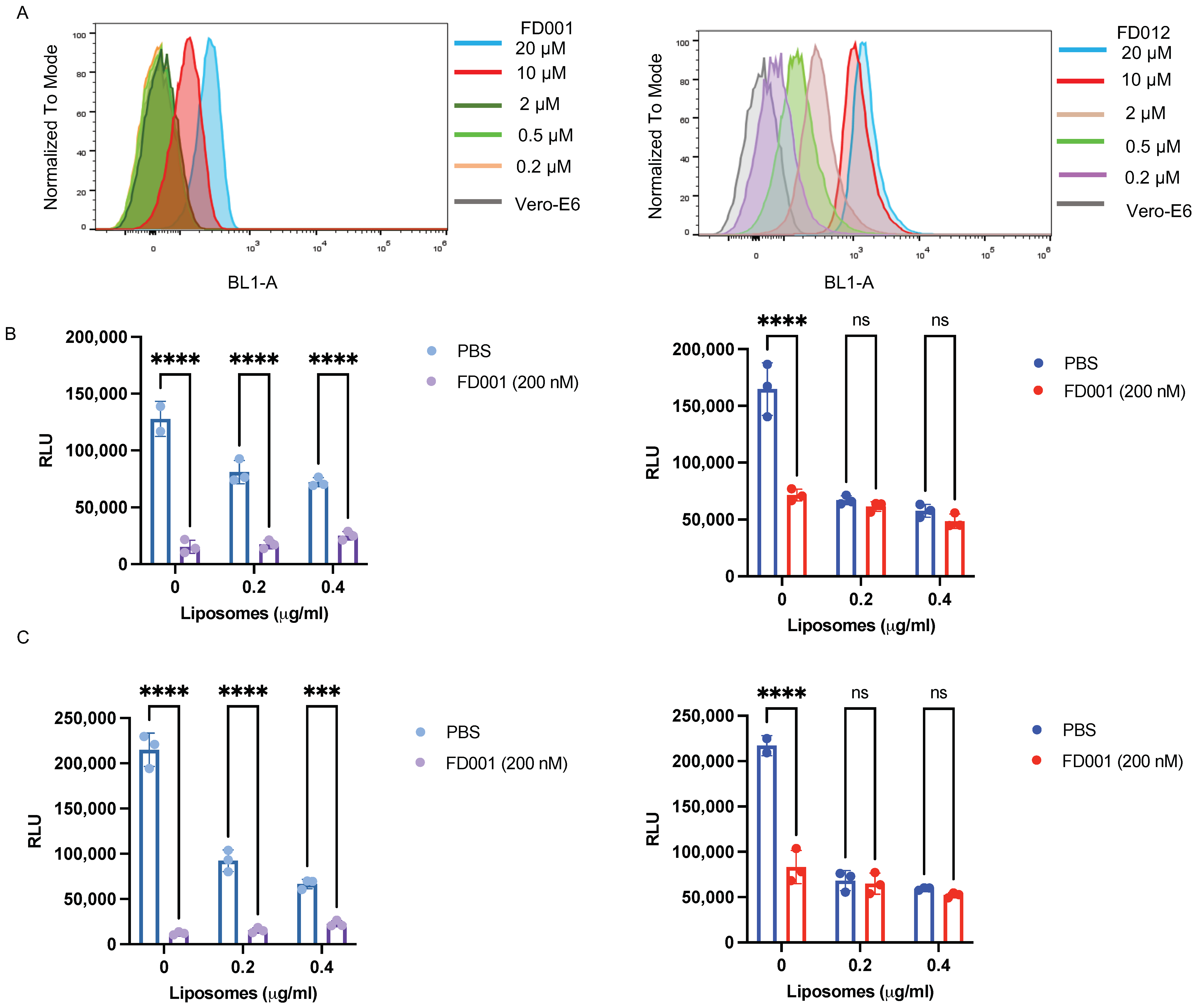
3.1. Inhibitory and Inactivating Activity of FD001 and Analogs on Infection of SARS-CoV-2 and Its Variants
3.2. Inhibitory and Inactivating Activity of FD-Compounds on SARS-CoV and MERS-CoV Infection
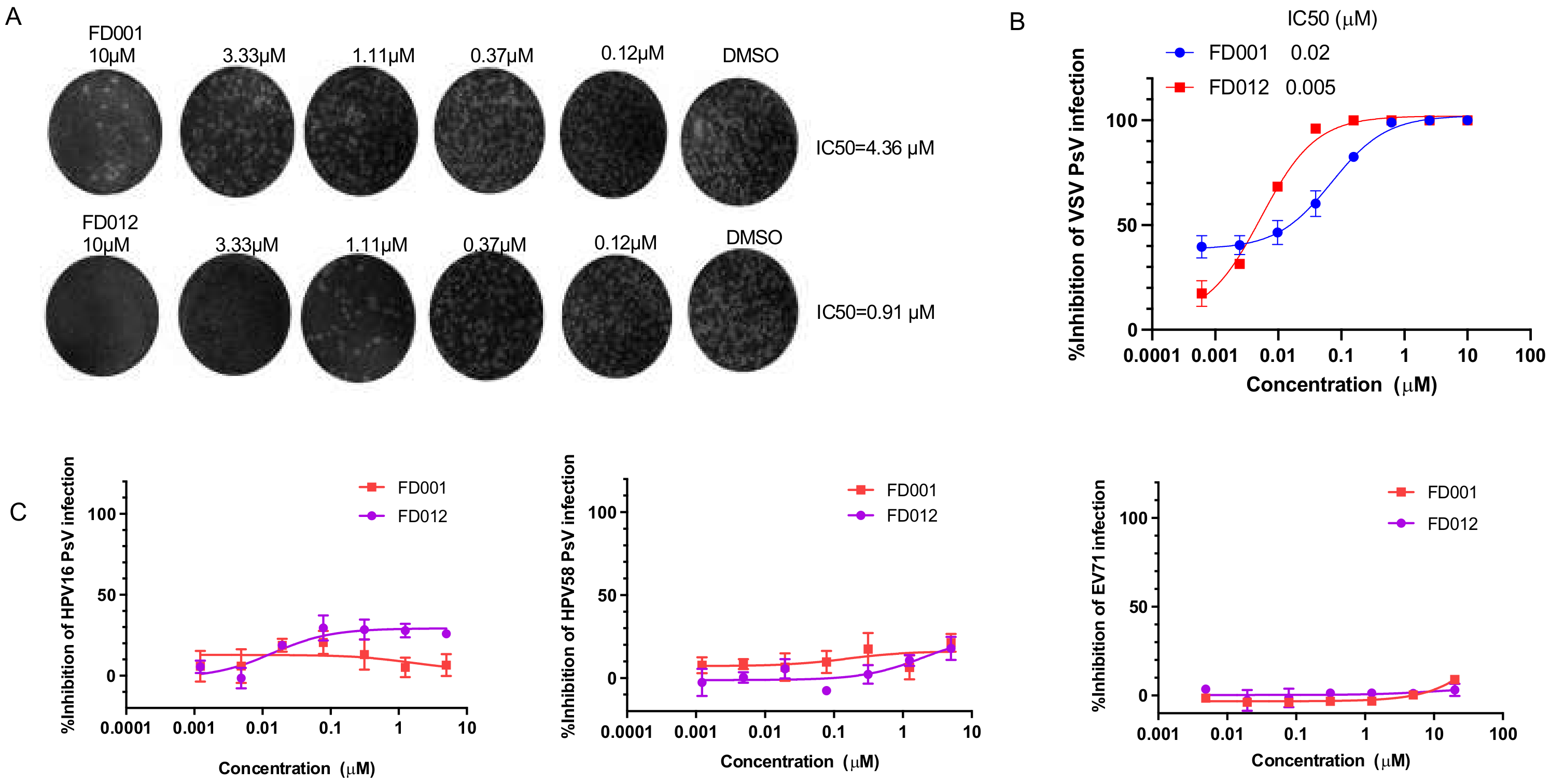
3.3. FD-Compounds Effectively Inhibited Infection by Other Enveloped Viruses with Class I Viral Fusion Proteins
3.4. FD-Compounds Effectively Inhibited Infection by Enveloped Viruses with Class II and III Viral Fusion Protein, but Not Nonenveloped Virus Infection
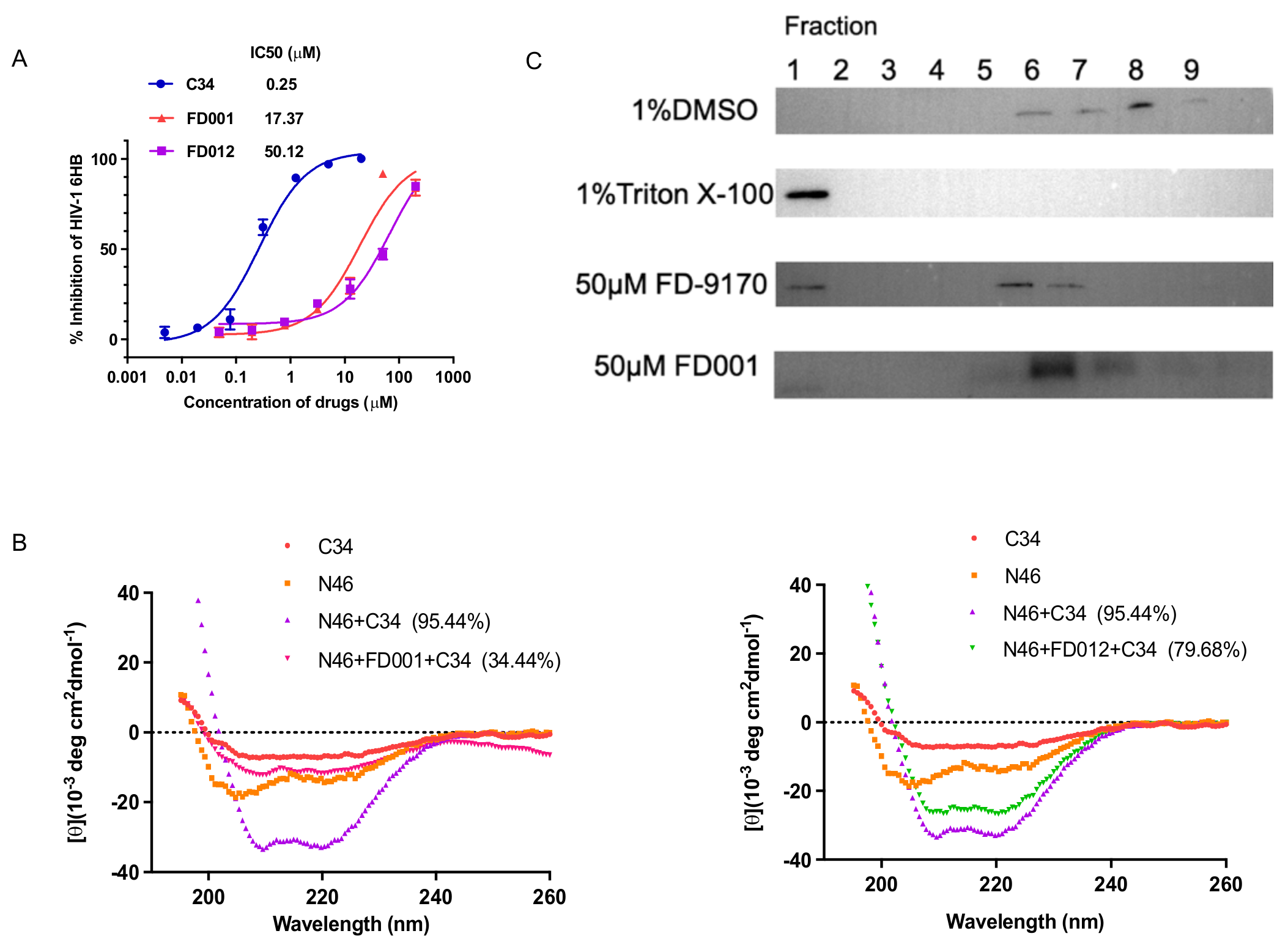
3.5. FD-Compounds Exerted Antiviral Effects through Multiple Mechanisms of Action
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, P.I.; Hsueh, P.R. Emerging threats from zoonotic coronaviruses-from SARS and MERS to 2019-nCoV. J. Microbiol. Immunol. Infect. 2020, 53, 365–367. [Google Scholar] [CrossRef]
- Ayittey, F.K.; Ayittey, M.K.; Chiwero, N.B.; Kamasah, J.S.; Dzuvor, C. Economic impacts of Wuhan 2019-nCoV on China and the world. J. Med. Virol. 2020, 92, 473–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kleber de Oliveira, W.; Cortez-Escalante, J.; De Oliveira, W.T.; do Carmo, G.M.; Henriques, C.M.; Coelho, G.E.; Araújo de França, G.V. Increase in Reported Prevalence of Microcephaly in Infants Born to Women Living in Areas with Confirmed Zika Virus Transmission During the First Trimester of Pregnancy—Brazil, 2015. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 242–247. [Google Scholar] [CrossRef]
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coltart, C.E.; Lindsey, B.; Ghinai, I.; Johnson, A.M.; Heymann, D.L. The Ebola outbreak, 2013–2016: Old lessons for new epidemics. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160297. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, Y.; Huang, B.; Deng, W.; Quan, Y.; Wang, W.; Xu, W.; Zhao, Y.; Li, N.; Zhang, J.; et al. Development of an Inactivated Vaccine Candidate, BBIBP-CorV, with Potent Protection against SARS-CoV-2. Cell 2020, 182, 713–721.e9. [Google Scholar] [CrossRef]
- Xia, S.; Zhang, Y.; Wang, Y.; Wang, H.; Yang, Y.; Gao, G.F.; Tan, W.; Wu, G.; Xu, M.; Lou, Z.; et al. Safety and immunogenicity of an inactivated SARS-CoV-2 vaccine, BBIBP-CorV: A randomised, double-blind, placebo-controlled, phase 1/2 trial. Lancet Infect. Dis. 2021, 21, 39–51. [Google Scholar] [CrossRef]
- Zhang, Y.; Zeng, G.; Pan, H.; Li, C.; Hu, Y.; Chu, K.; Han, W.; Chen, Z.; Tang, R.; Yin, W.; et al. Safety, tolerability, and immunogenicity of an inactivated SARS-CoV-2 vaccine in healthy adults aged 18–59 years: A randomised, double-blind, placebo-controlled, phase 1/2 clinical trial. Lancet Infect. Dis. 2021, 21, 181–192. [Google Scholar] [CrossRef]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416. [Google Scholar] [CrossRef]
- Zhu, F.C.; Guan, X.H.; Li, Y.H.; Huang, J.Y.; Jiang, T.; Hou, L.H.; Li, J.X.; Yang, B.F.; Wang, L.; Wang, W.J.; et al. Immunogenicity and safety of a recombinant adenovirus type-5-vectored COVID-19 vaccine in healthy adults aged 18 years or older: A randomised, double-blind, placebo-controlled, phase 2 trial. Lancet 2020, 396, 479–488. [Google Scholar] [CrossRef]
- Ramasamy, M.N.; Minassian, A.M.; Ewer, K.J.; Flaxman, A.L.; Folegatti, P.M.; Owens, D.R.; Voysey, M.; Aley, P.K.; Angus, B.; Babbage, G.; et al. Safety and immunogenicity of ChAdOx1 nCoV-19 vaccine administered in a prime-boost regimen in young and old adults (COV002): A single-blind, randomised, controlled, phase 2/3 trial. Lancet 2021, 396, 1979–1993. [Google Scholar] [CrossRef]
- Sadoff, J.; Le Gars, M.; Shukarev, G.; Heerwegh, D.; Truyers, C.; de Groot, A.M.; Stoop, J.; Tete, S.; Van Damme, W.; Leroux-Roels, I.; et al. Interim Results of a Phase 1-2a Trial of Ad26.COV2.S COVID-19 Vaccine. N. Engl. J. Med. 2021, 384, 1824–1835. [Google Scholar] [CrossRef]
- Lamb, Y.N. Remdesivir: First Approval. Drugs 2020, 80, 1355–1363. [Google Scholar] [CrossRef]
- Holman, W.; Holman, W.; McIntosh, S.; Painter, W.; Painter, G.; Bush, J.; Cohen, O. Accelerated first-in-human clinical trial of EIDD-2801/MK-4482 (molnupiravir), a ribonucleoside analog with potent antiviral activity against SARS-CoV-2. Trials 2021, 22, 561. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Tala, S.R.; Lu, H.; Vakulenko, A.V.; Chen, Q.Y.; Sivapackiam, J.; Pandya, K.; Jiang, S.; Debnath, A.K. Design, synthesis, and structure-activity relationship of a novel series of 2-aryl 5-(4-oxo-3-phenethyl-2-thioxothiazolidinylidenemethyl)furans as HIV-1 entry inhibitors. J. Med. Chem. 2009, 52, 7631–7639. [Google Scholar] [CrossRef] [Green Version]
- Hua, C.; Zhu, Y.; Wu, C.; Si, L.; Wang, Q.; Sui, L.; Jiang, S. The Underlying Mechanism of 3-Hydroxyphthalic Anhydride-Modified Bovine Beta-Lactoglobulin to Block Human Papillomavirus Entry Into the Host Cell. Front. Microbiol. 2019, 10, 2188. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, C.; Chen, B.; Wang, Q.; Xu, W.; Ye, S.; Jiang, S.; Zhu, Y.; Zhang, R. Crystal Structure of Refolding Fusion Core of Lassa Virus GP2 and Design of Lassa Virus Fusion Inhibitors. Front. Microbiol. 2019, 10, 1829. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Hua, C.; Xia, S.; Li, W.; Lu, L.; Jiang, S. Combining a Fusion Inhibitory Peptide Targeting the MERS-CoV S2 Protein HR1 Domain and a Neutralizing Antibody Specific for the S1 Protein Receptor-Binding Domain (RBD) Showed Potent Synergism against Pseudotyped MERS-CoV with or without Mutations in RBD. Viruses 2019, 11, 31. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Xia, S.; Pu, J.; Wang, Q.; Li, P.; Lu, L.; Jiang, S. The Antihistamine Drugs Carbinoxamine Maleate and Chlorpheniramine Maleate Exhibit Potent Antiviral Activity Against a Broad Spectrum of Influenza Viruses. Front. Microbiol. 2018, 9, 2643. [Google Scholar] [CrossRef]
- Gao, Y.; Tai, W.; Wang, N.; Li, X.; Jiang, S.; Debnath, A.K.; Du, L.; Chen, S. Identification of Novel Natural Products as Effective and Broad-Spectrum Anti-Zika Virus Inhibitors. Viruses 2019, 11, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Liu, M.; Wang, C.; Xu, W.; Lan, Q.; Feng, S.; Qi, F.; Bao, L.; Du, L.; Liu, S.; et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020, 30, 343–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, Q.; Wang, Q.; Chen, W.; Du, L.; Dimitrov, D.S.; Lu, L.; Jiang, S. HIV-1 gp41-targeting fusion inhibitory peptides enhance the gp120-targeting protein-mediated inactivation of HIV-1 virions. Emerg. Microbes Infect. 2017, 6, e59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pu, J.; Dai, Y.; Wang, Q.; Lu, L.; Zhang, J.; Xu, W.; Xie, L.; Wang, S.; Yu, F.; He, X.; et al. Rational Design of A Novel Small-Molecule HIV-1 Inactivator Targeting Both gp120 and gp41 of HIV-1. Front. Pharmacol. 2020, 11, 613361. [Google Scholar] [CrossRef]
- Su, S.; Rasquinha, G.; Du, L.; Wang, Q.; Xu, W.; Li, W.; Lu, L.; Jiang, S. A Peptide-Based HIV-1 Fusion Inhibitor with Two Tail-Anchors and Palmitic Acid Exhibits Substantially Improved In Vitro and Ex Vivo Anti-HIV-1 Activity and Prolonged In Vivo Half-Life. Molecules 2019, 24, 1134. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Su, S.; Xue, J.; Yu, F.; Pu, J.; Bi, W.; Xia, S.; Meng, Y.; Wang, C.; Yang, W.; et al. An amphipathic peptide targeting the gp41 cytoplasmic tail kills HIV-1 virions and infected cells. Sci. Transl. Med. 2020, 12, eaaz2254. [Google Scholar] [CrossRef]
- Drosten, C.; Günther, S.; Preiser, W.; van der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef]
- Zaki, A.M.; van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Lu, H.; Siddiqui, P.; Zhou, Y.; Jiang, S. Receptor-binding domain of severe acute respiratory syndrome coronavirus spike protein contains multiple conformation-dependent epitopes that induce highly potent neutralizing antibodies. J. Immunol. 2005, 174, 4908–4915. [Google Scholar] [CrossRef] [Green Version]
- Cheng, G.; Montero, A.; Gastaminza, P.; Whitten-Bauer, C.; Wieland, S.F.; Isogawa, M.; Fredericksen, B.; Selvarajah, S.; Gallay, P.A.; Ghadiri, M.R.; et al. A virocidal amphipathic {alpha}-helical peptide that inhibits hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2008, 105, 3088–3093. [Google Scholar] [CrossRef] [Green Version]
- Wolf, M.C.; Freiberg, A.N.; Zhang, T.; Akyol-Ataman, Z.; Grock, A.; Hong, P.W.; Li, J.; Watson, N.F.; Fang, A.Q.; Aguilar, H.C.; et al. A broad-spectrum antiviral targeting entry of enveloped viruses. Proc. Natl. Acad. Sci. USA 2010, 107, 3157–3162. [Google Scholar] [CrossRef] [Green Version]
- Hollmann, A.; Castanho, M.A.; Lee, B.; Santos, N.C. Singlet oxygen effects on lipid membranes: Implications for the mechanism of action of broad-spectrum viral fusion inhibitors. Biochem. J. 2014, 459, 161–170. [Google Scholar] [CrossRef]
- Vigant, F.; Lee, J.; Hollmann, A.; Tanner, L.B.; Akyol Ataman, Z.; Yun, T.; Shui, G.; Aguilar, H.C.; Zhang, D.; Meriwether, D.; et al. A mechanistic paradigm for broad-spectrum antivirals that target virus-cell fusion. PLoS Pathog. 2013, 9, e1003297. [Google Scholar] [CrossRef]
- Khursheed, A.; Jain, V.; Rasool, A.; Rather, M.A.; Malik, N.A.; Shalla, A.H. Molecular scaffolds from mother nature as possible lead compounds in drug design and discovery against coronaviruses: A landscape analysis of published literature and molecular docking studies. Microb. Pathog. 2021, 157, 104933. [Google Scholar] [CrossRef]
- Tariq, S.; Wani, S.; Rasool, W.; Shafi, K.; Bhat, M.A.; Prabhakar, A.; Shalla, A.H.; Rather, M.A. A comprehensive review of the antibacterial, antifungal and antiviral potential of essential oils and their chemical constituents against drug-resistant microbial pathogens. Microb. Pathog. 2019, 134, 103580. [Google Scholar] [CrossRef]
- Highleyman, L. FDA approves fomivirsen, famciclovir, and Thalidomide. Food and Drug Administration. Beta 1998, 5. [Google Scholar] [PubMed]
- Baker, R. FDA approves oral ganciclovir as first drug to prevent CMV disease. Food and Drug Administration. Beta 1995, 8. [Google Scholar] [PubMed]
- Schouten, J.T. Oral ganciclovir. STEP Perspect 1995, 7, 1–11. [Google Scholar]
- Zidovudine approved by FDA for treatment of AIDS. Clin. Pharm. 1987, 6, 431–435.
- Friedland, G. FDA approves d4T as alternative to AZT, ddI, or ddC. Food and Drug Administration. AIDS Clin. Care 1995, 7, 4–10. [Google Scholar]
- Li, R.; Hou, Y.; Huang, J.; Pan, W.; Ma, Q.; Shi, Y.; Li, C.; Zhao, J.; Jia, Z.; Jiang, H.; et al. Lianhuaqingwen exerts anti-viral and anti-inflammatory activity against novel coronavirus (SARS-CoV-2). Pharmacol. Res. 2020, 156, 104761. [Google Scholar] [CrossRef]
- Nair, M.S.; Huang, Y.; Fidock, D.A.; Polyak, S.J.; Wagoner, J.; Towler, M.J.; Weathers, P.J. Artemisia annua L. extracts inhibit the in vitro replication of SARS-CoV-2 and two of its variants. J. Ethnopharmacol. 2021, 274, 114016. [Google Scholar] [CrossRef]
- Bailly, C.; Vergoten, G. Glycyrrhizin: An alternative drug for the treatment of COVID-19 infection and the associated respiratory syndrome? Pharmacol. Ther. 2020, 214, 107618. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | R | MW(Da) |
|---|---|---|
| FD001 | H | 470.0 |
| FD007 |  | 524.01 |
| FD008 |  | 568.06 |
| FD009 |  | 514.01 |
| FD010 |  | 594.07 |
| FD012 |  | 540.05 |
| FD013 |  | 582.11 |
| Compound | CC50 (µM) | IC50 (µM) | SI | EC50 (µM) | SI |
|---|---|---|---|---|---|
| CC50/IC50 | CC50/EC50 | ||||
| SARS-CoV PsV infection in hACE2-293T cells | |||||
| FD001 | 28.84 ± 1.24 | 0.18 ± 0.02 | 160.22 | 1.58 ± 0.21 | 18.25 |
| FD007 | 33.11 ± 1.02 | 0.11 ± 0.01 | 301.00 | 1.54 ± 0.14 | 21.50 |
| FD008 | 23.44 ± 0.78 | 0.05 ± 0.01 | 468.80 | 0.19 ± 0.02 | 123.37 |
| FD009 | 17.37 ± 0.64 | 0.03 ± 0.01 | 579.00 | 0.12 ± 0.01 | 144.75 |
| FD010 | 20.42 ± 1.00 | 0.29 ± 0.03 | 70.41 | 0.19 ± 0.03 | 107.47 |
| FD012 | 12.30 ± 0.34 | 0.02 ± 0.01 | 615.00 | 0.12 ± 0.01 | 102.50 |
| FD013 | 18.62 ± 0.23 | 0.05 ± 0.01 | 372.40 | 0.10 ± 0.01 | 186.20 |
| MERS-CoV PsV infection in Huh-7 cells | |||||
| FD001 | 42.65 ± 1.31 | 0.29 ± 0.03 | 147.07 | 1.12 ± 0.16 | 38.08 |
| FD007 | >50 | 0.20 ± 0.03 | >250 | 0.89 ± 0.09 | >56.18 |
| FD008 | 38.90 ± 1.22 | 0.31 ± 0.02 | 125.48 | 0.69 ± 0.13 | 56.38 |
| FD009 | 27.54 ± 1.43 | 0.28 ± 0.02 | 98.36 | 0.29 ± 0.02 | 94.97 |
| FD010 | 44.67 ± 1.26 | 0.12 ± 0.01 | 372.25 | 0.04 ± 0.01 | 1116.75 |
| FD012 | 18.62 ± 0.35 | 0.11 ± 0.01 | 169.27 | 0.05 ± 0.01 | 372.40 |
| FD013 | 30.90 ± 0.63 | 0.05 ± 0.01 | 618.00 | 0.03 ± 0.01 | 1030.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pu, J.; He, X.; Xu, W.; Wang, C.; Lan, Q.; Hua, C.; Wang, K.; Lu, L.; Jiang, S. The Analogs of Furanyl Methylidene Rhodanine Exhibit Broad-Spectrum Inhibitory and Inactivating Activities against Enveloped Viruses, including SARS-CoV-2 and Its Variants. Viruses 2022, 14, 489. https://doi.org/10.3390/v14030489
Pu J, He X, Xu W, Wang C, Lan Q, Hua C, Wang K, Lu L, Jiang S. The Analogs of Furanyl Methylidene Rhodanine Exhibit Broad-Spectrum Inhibitory and Inactivating Activities against Enveloped Viruses, including SARS-CoV-2 and Its Variants. Viruses. 2022; 14(3):489. https://doi.org/10.3390/v14030489
Chicago/Turabian StylePu, Jing, Xiaoyang He, Wei Xu, Cong Wang, Qiaoshuai Lan, Chen Hua, Kai Wang, Lu Lu, and Shibo Jiang. 2022. "The Analogs of Furanyl Methylidene Rhodanine Exhibit Broad-Spectrum Inhibitory and Inactivating Activities against Enveloped Viruses, including SARS-CoV-2 and Its Variants" Viruses 14, no. 3: 489. https://doi.org/10.3390/v14030489
APA StylePu, J., He, X., Xu, W., Wang, C., Lan, Q., Hua, C., Wang, K., Lu, L., & Jiang, S. (2022). The Analogs of Furanyl Methylidene Rhodanine Exhibit Broad-Spectrum Inhibitory and Inactivating Activities against Enveloped Viruses, including SARS-CoV-2 and Its Variants. Viruses, 14(3), 489. https://doi.org/10.3390/v14030489