
Targeting of Protein Kinase CK2 Elicits Antiviral Activity on Bovine Coronavirus Infection

 ,
,  , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and BCoV Virus
2.2. Compounds
2.3. Cell Cytotoxicity Assay and Drug Treatments
2.4. Cytopathic Effect Assay
2.5. Crystal Violet Assay
2.6. Plaque Reduction Assay
2.7. Quantitative Real-Time PCR Assays
2.8. BCoV Viral Proteins Detection by Western Blot
2.9. BCoV Viral Proteins Detection by Immunocytochemistry
2.10. Pull-Down Assay
2.11. LC-MS/MS Analysis and Protein Identification
2.12. Confocal Microscopy
2.13. CK2 Signaling Experiments
2.14. Statistical Analysis
3. Results
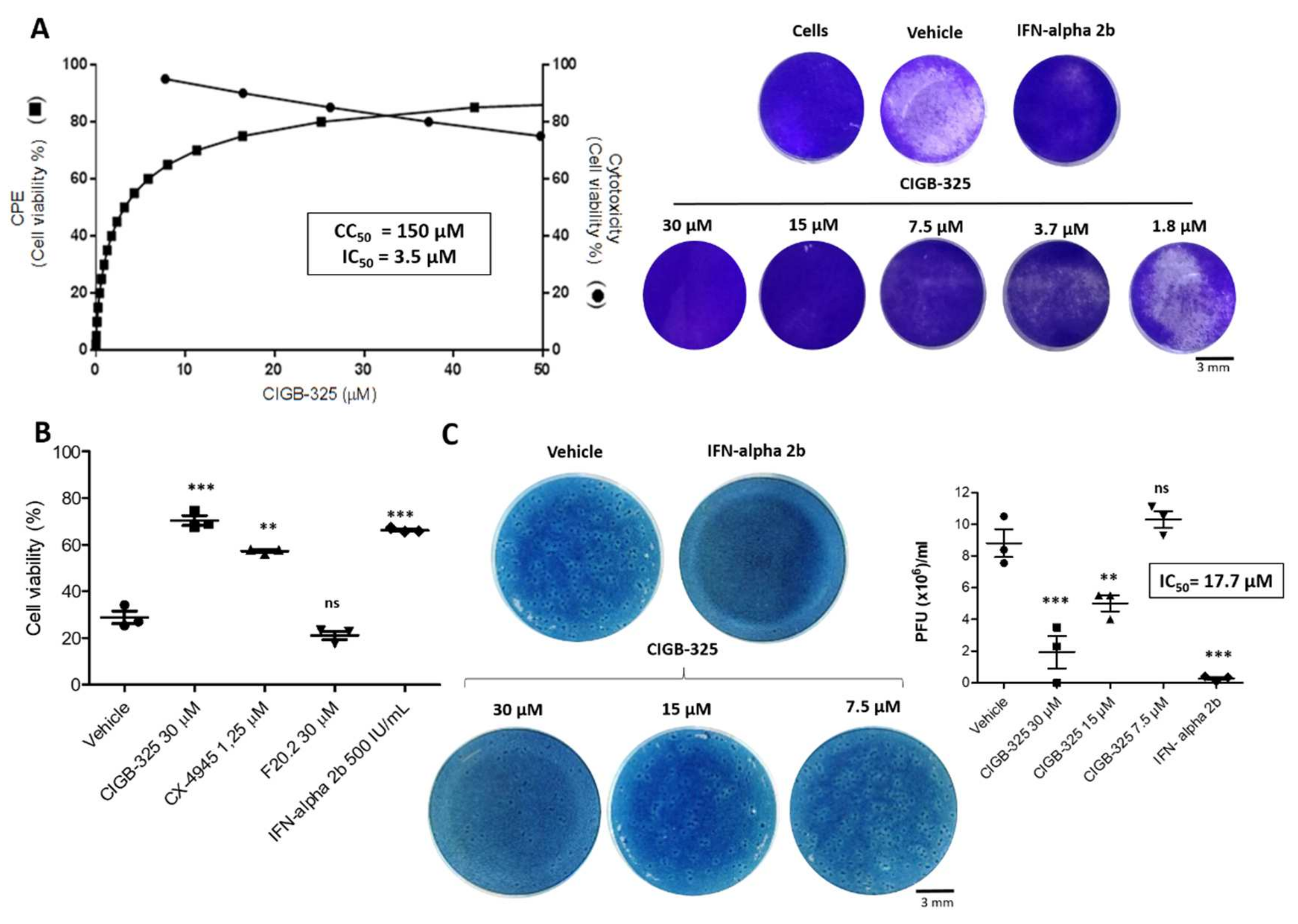
3.1. CIGB-325 Exhibits Antiviral Effect on Bovine Coronavirus Infected Cells
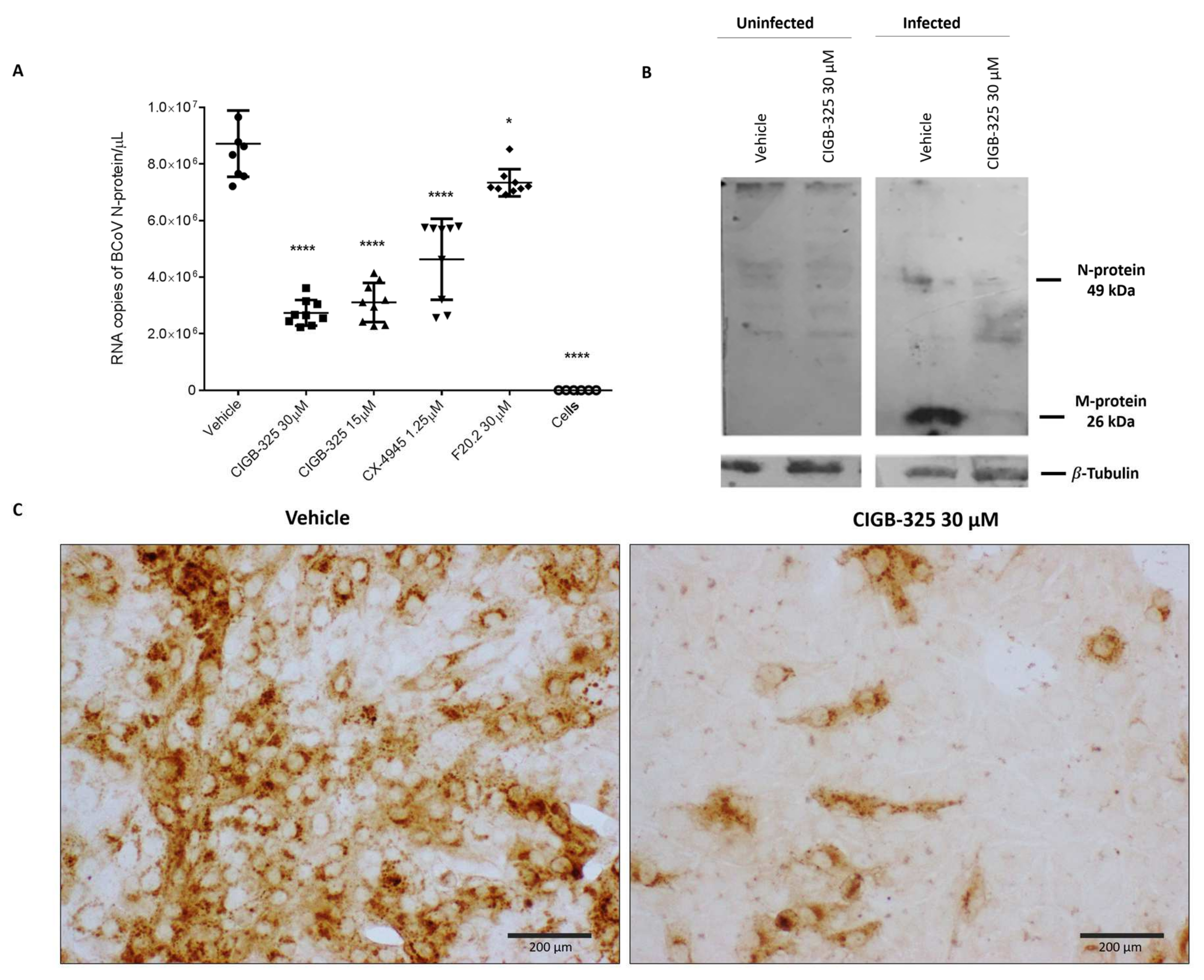
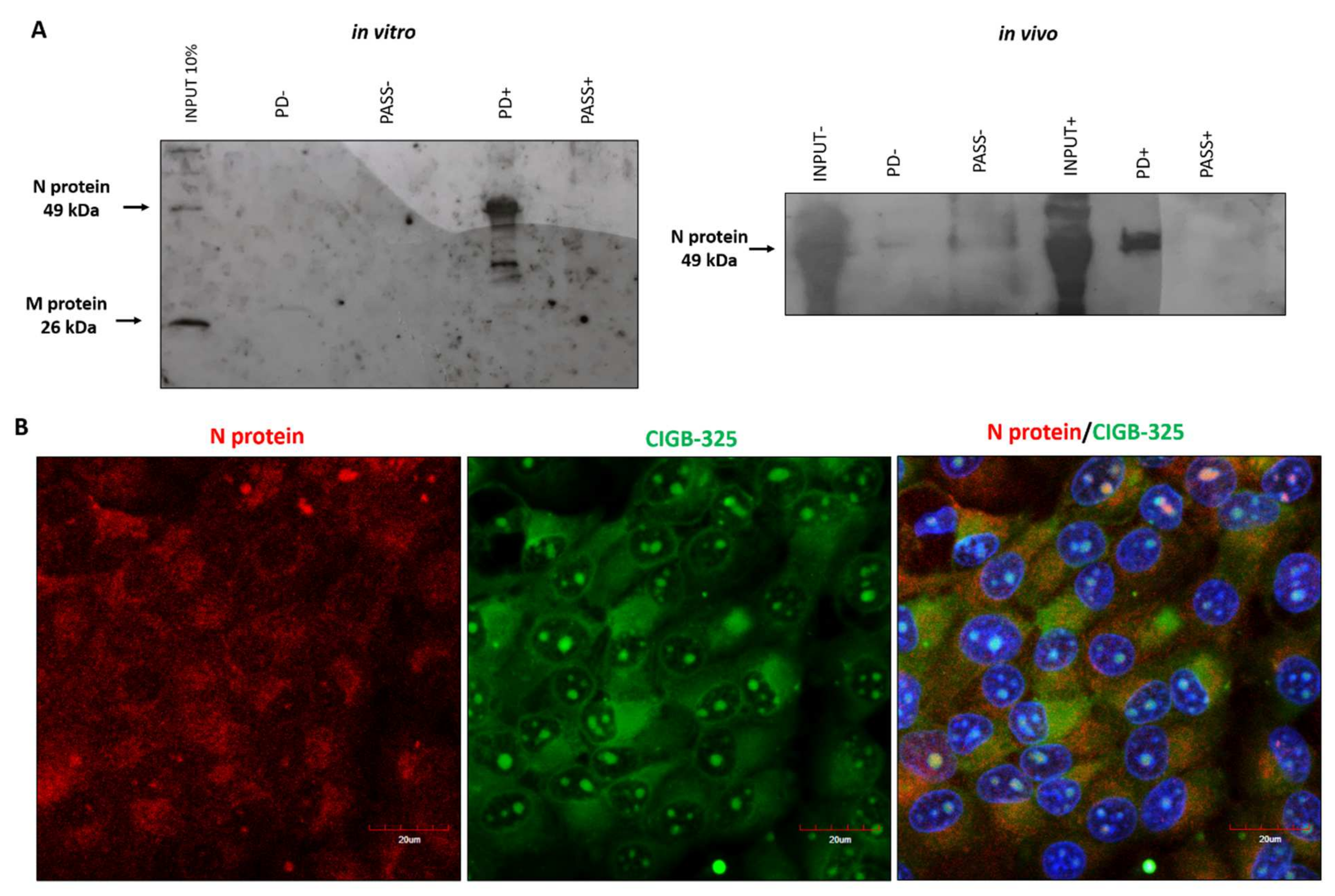
3.2. CIGB-325 Targets BCoV N Protein in MDBK Cells
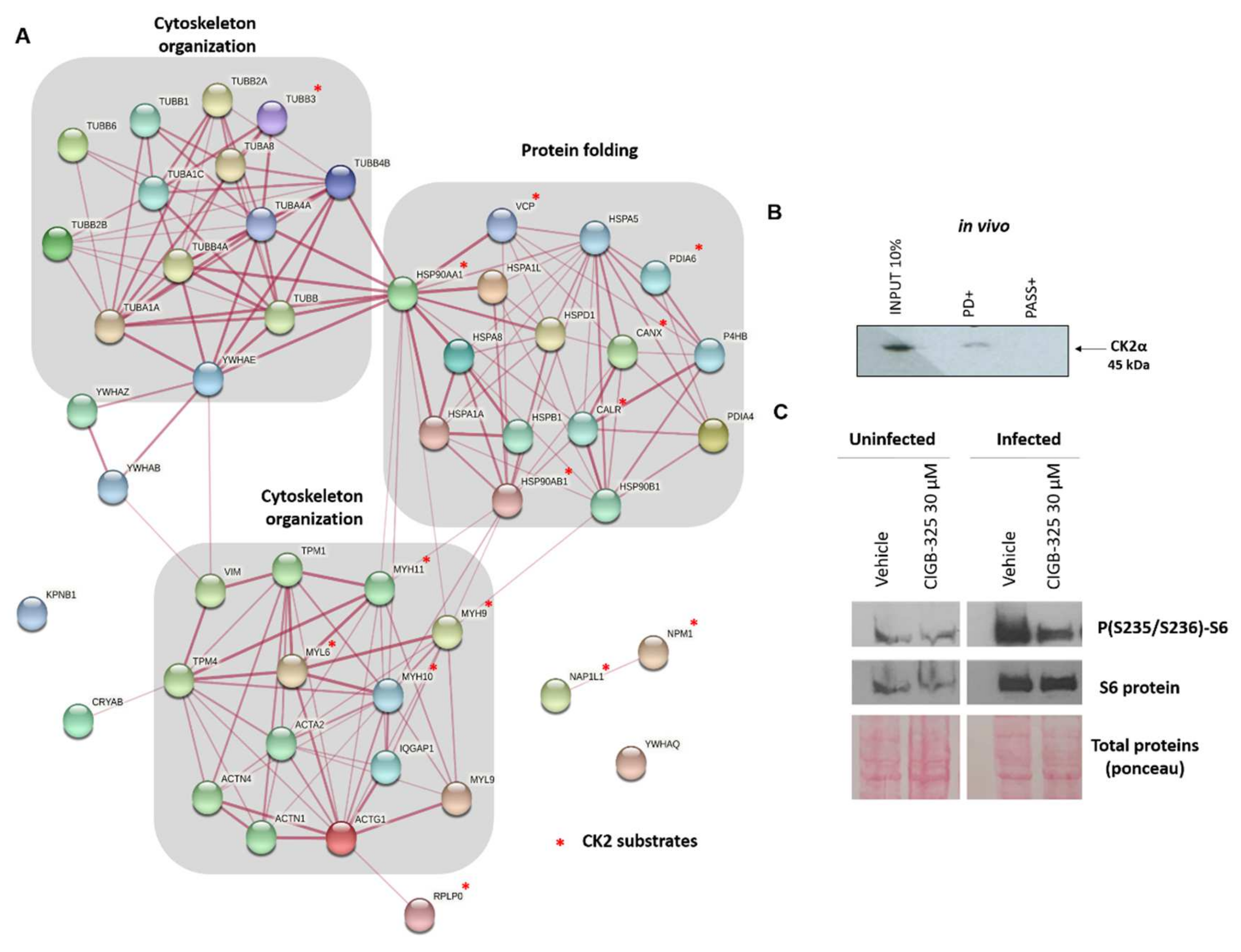
3.3. CIGB-325 Interactomic Landscape in BCoV-Mebus-Infected MDBK Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Masters, P.S. The molecular biology of coronaviruses. Adv. Virus Res. 2006, 66, 193–292. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.R.; Leibowitz, J.L. Coronavirus pathogenesis. Adv. Virus Res. 2011, 81, 85–164. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.R.; Navas-Martin, S. Coronavirus pathogenesis and the emerging pathogen severe acute respiratory syndrome coronavirus. Microbiol. Mol. Biol. Rev. MMBR 2005, 69, 635–664. [Google Scholar] [CrossRef] [Green Version]
- Peiris, J.S.; Guan, Y.; Yuen, K.Y. Severe acute respiratory syndrome. Nat. Med. 2004, 10 (Suppl. 12), S88–S97. [Google Scholar] [CrossRef] [PubMed]
- Azhar, E.I.; Hui, D.S.C.; Memish, Z.A.; Drosten, C.; Zumla, A. The Middle East Respiratory Syndrome (MERS). Infect. Dis. Clin. North. Am. 2019, 33, 891–905. [Google Scholar] [CrossRef]
- Saif, L.J. Animal coronavirus vaccines: Lessons for SARS. Dev. Biol. 2004, 119, 129–140. [Google Scholar]
- Pushpakom, S.; Iorio, F.; Eyers, P.A.; Escott, K.J.; Hopper, S.; Wells, A.; Doig, A.; Guilliams, T.; Latimer, J.; McNamee, C.; et al. Drug repurposing: Progress, challenges and recommendations. Nat. Reviews. Drug Discov. 2019, 18, 41–58. [Google Scholar] [CrossRef]
- Skelton, D.J.; Alsobhe, A.; Anastasi, E.; Atallah, C.; Bird, J.; Brown, B.; Didon, D.; Gater, P.; James, K.; Lennon, D.D., Jr. Drug repurposing prediction for COVID-19 using probabilistic networks and crowdsourced curation. Arxiv Prepr. 2020, arXiv:2005.11088. [Google Scholar]
- Prussia, A.; Thepchatri, P.; Snyder, J.P.; Plemper, R.K. Systematic approaches towards the development of host-directed antiviral therapeutics. Int. J. Mol. Sci. 2011, 12, 4027–4052. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Hiatt, J.; Bouhaddou, M.; Rezelj, V.V.; Ulferts, S.; Braberg, H.; Jureka, A.S.; Obernier, K.; Guo, J.Z.; Batra, J.; et al. Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science 2020, 370, eabe9403. [Google Scholar] [CrossRef]
- Keating, J.A.; Striker, R. Phosphorylation events during viral infections provide potential therapeutic targets. Rev. Med. Virol. 2012, 22, 166–181. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The global phosphorylation landscape of SARS-CoV-2 infection. Cell 2020, 182, 685–712.e19. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Dickmander, R.J.; Bayati, A.; Taft-Benz, S.A.; Smith, J.L.; Brown, J.W.; Lenarcic, E.M.; Yount, B.L.; Chang, E.; Axtman, A.D.; et al. Host kinase CSNK2 is a target for inhibition of pathogenic β-coronaviruses including SARS-CoV-2. bioRxiv 2022. [Google Scholar] [CrossRef]
- Perea, S.E.; Reyes, O.; Puchades, Y.; Mendoza, O.; Vispo, N.S.; Torrens, I.; Santos, A.; Silva, R.; Acevedo, B.; López, E.; et al. Antitumor effect of a novel proapoptotic peptide that impairs the phosphorylation by the protein kinase 2 (casein kinase 2). Cancer Res. 2004, 64, 7127–7129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, Y.; Melão, A.; Ramón, A.C.; Vázquez, D.; Ribeiro, D.; Perea, S.E.; Barata, J.T. Clinical-Grade peptide-based inhibition of CK2 blocks viability and proliferation of T-ALL cells and counteracts IL-7 stimulation and stromal support. Cancers 2020, 12, 1377. [Google Scholar] [CrossRef] [PubMed]
- Perera, Y.; Ramos, Y.; Padrón, G.; Caballero, E.; Guirola, O.; Caligiuri, L.G.; Lorenzo, N.; Gottardo, F.; Farina, H.G.; Filhol, O.; et al. CIGB-300 anticancer peptide regulates the protein kinase CK2-dependent phosphoproteome. Mol. Cell. Biochem. 2020, 470, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Águila, J.D.F.; Vega, Y.G.; Jiménez, R.O.R.; Sacerio, A.L.; Rodríguez, C.R.R.; Fraga, Y.R.; Silva, C.V. Safety of intravenous application of cigb-300 in patients with hematological malignancies. EHPMA study. Rev. Cuba. Hematol. Inmunol. Hemoter. 2016, 32, 236–248. [Google Scholar]
- García-Diegues, R.; de la Torre-Santos, A. Phase I Study of CIGB-300 administered intravenously in patients with relapsed/refractory solid tumors. Arch. Med. 2018, 1, 4. [Google Scholar]
- Nouri, K.; Moll, J.M.; Milroy, L.G.; Hain, A.; Dvorsky, R.; Amin, E.; Lenders, M.; Nagel-Steger, L.; Howe, S.; Smits, S.H.; et al. Biophysical characterization of nucleophosmin interactions with human immunodeficiency virus rev and herpes simplex virus US11. PLoS ONE 2015, 10, e0143634. [Google Scholar] [CrossRef] [Green Version]
- Cruz, L.R.; Baladrón, I.; Rittoles, A.; Díaz, P.A.; Valenzuela, C.; Santana, R.; Vázquez, M.M.; García, A.; Chacón, D.; Thompson, D.; et al. Treatment with an Anti-CK2 synthetic peptide improves clinical response in COVID-19 Patients with pneumonia. A randomized and controlled clinical trial. ACS Pharmacol. Transl. Sci. 2021, 4, 206–212. [Google Scholar] [CrossRef]
- Kenney, S.P.; Wang, Q.; Vlasova, A.; Jung, K.; Saif, L. Naturally occurring animal coronaviruses as models for studying highly pathogenic human coronaviral disease. Vet. Pathol. 2021, 58, 438–452. [Google Scholar] [CrossRef] [PubMed]
- Alluwaimi, A.M.; Alshubaith, I.H.; Al-Ali, A.M.; Abohelaika, S. The coronaviruses of animals and birds: Their zoonosis, vaccines, and models for SARS-CoV and SARS-CoV2. Front. Vet. Sci. 2020, 7, 582287. [Google Scholar] [CrossRef] [PubMed]
- Saif, L.J. Bovine respiratory coronavirus. Vet. Clin. North. America. Food Anim. Pract. 2010, 26, 349–364. [Google Scholar] [CrossRef] [PubMed]
- Vlasova, A.N.; Saif, L.J. Bovine coronavirus and the associated diseases. Front. Vet. Sci. 2021, 8, 643220. [Google Scholar] [CrossRef]
- Zhang, X.; Hasoksuz, M.; Spiro, D.; Halpin, R.; Wang, S.; Vlasova, A.; Janies, D.; Jones, L.R.; Ghedin, E.; Saif, L.J. Quasispecies of bovine enteric and respiratory coronaviruses based on complete genome sequences and genetic changes after tissue culture adaptation. Virology 2007, 363, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. Clifton N.J. 2015, 1282, 1–23. [Google Scholar] [CrossRef] [Green Version]
- Perrin-Cocon, L.; Diaz, O.; Jacquemin, C.; Barthel, V.; Ogire, E.; Ramière, C.; André, P.; Lotteau, V.; Vidalain, P.O. The current landscape of coronavirus-host protein-protein interactions. J. Transl. Med. 2020, 18, 319. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Horgan, G.W.; Dempfle, L. Relative expression software tool (REST) for group-wise comparison and statistical analysis of relative expression results in real-time PCR. Nucleic Acids Res. 2002, 30, e36. [Google Scholar] [CrossRef]
- Ruijter, J.M.; Ramakers, C.; Hoogaars, W.M.; Karlen, Y.; Bakker, O.; van den Hoff, M.J.; Moorman, A.F. Amplification efficiency: Linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 2009, 37, e45. [Google Scholar] [CrossRef] [Green Version]
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meggio, F.; Pinna, L.A. One-thousand-and-one substrates of protein kinase CK2? FASEB J. 2003, 17, 349–368. [Google Scholar] [CrossRef] [PubMed]
- Bian, Y.; Ye, M.; Wang, C.; Cheng, K.; Song, C.; Dong, M.; Pan, Y.; Qin, H.; Zou, H. Global screening of CK2 kinase substrates by an integrated phosphoproteomics workflow. Sci. Rep. 2013, 3, 3460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, D.; Garcia, C.; Gonzales, M.; Garay, H.; Diago, D.; Guzman, L.; Ferro, W.; Quintana, M.; Gomez, L.; Chavez, B.; et al. Monoclonal and polyclonal antibodies as biological reagents for SARS-CoV-2 diagnosis though nucleocapsid protein detection. BioProcess. J. 2021, 20. [Google Scholar] [CrossRef]
- Surjit, M.; Kumar, R.; Mishra, R.N.; Reddy, M.K.; Chow, V.T.; Lal, S.K. The severe acute respiratory syndrome coronavirus nucleocapsid protein is phosphorylated and localizes in the cytoplasm by 14-3-3-mediated translocation. J. Virol. 2005, 79, 11476–11486. [Google Scholar] [CrossRef] [Green Version]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Perea, S.E.; Baladron, I.; Garcia, Y.; Perera, Y.; Lopez, A.; Soriano, J.L.; Batista, N.; Palau, A.; Hernández, I.; Farina, H.; et al. CIGB-300, a synthetic peptide-based drug that targets the CK2 phosphoaceptor domain. Translational and clinical research. Mol. Cell. Biochem. 2011, 356, 45–50. [Google Scholar] [CrossRef]
- Rosales, M.; Pérez, G.V.; Ramón, A.C.; Cruz, Y.; Rodríguez-Ulloa, A.; Besada, V.; Ramos, Y.; Vázquez-Blomquist, D.; Caballero, E.; Aguilar, D.; et al. Targeting of protein kinase CK2 in Acute myeloid leukemia cells using the clinical-grade synthetic-peptide CIGB-300. Biomedicines 2021, 9, 766. [Google Scholar] [CrossRef]
- Leung, W.K.; To, K.F.; Chan, P.K.; Chan, H.L.; Wu, A.K.; Lee, N.; Yuen, K.Y.; Sung, J.J. Enteric involvement of severe acute respiratory syndrome-associated coronavirus infection. Gastroenterology 2003, 125, 1011–1017. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Xu, Y.; Gao, R.; Lu, R.; Han, K.; Wu, G.; Tan, W. Detection of SARS-CoV-2 in different types of clinical specimens. Jama 2020, 323, 1843–1844. [Google Scholar] [CrossRef] [Green Version]
- Perera, Y.; Toro, N.D.; Gorovaya, L.; Fernandez, D.E.C.J.; Farina, H.G.; Perea, S.E. Synergistic interactions of the anti-casein kinase 2 CIGB-300 peptide and chemotherapeutic agents in lung and cervical preclinical cancer models. Mol. Clin. Oncol. 2014, 2, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beidas, M.; Chehadeh, W. Effect of human coronavirus OC43 Structural and accessory proteins on the transcriptional activation of antiviral response elements. Intervirology 2018, 61, 30–35. [Google Scholar] [CrossRef]
- Paladino, L.; Vitale, A.M.; Caruso Bavisotto, C.; Conway de Macario, E.; Cappello, F.; Macario, A.J.L.; Gammazza, A.M. The role of molecular chaperones in virus infection and implications for understanding and treating COVID-19. J. Clin. Med. 2020, 9, 3518. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhang, Y.; Lin, Z.; Shi, K.; Jiu, Y. Cytoskeleton-a crucial key in host cell for coronavirus infection. J. Mol. Cell Biol. 2021, 12, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Perera, Y.; Farina, H.G.; Gil, J.; Rodriguez, A.; Benavent, F.; Castellanos, L.; Gómez, R.E.; Acevedo, B.E.; Alonso, D.F.; Perea, S.E. Anticancer peptide CIGB-300 binds to nucleophosmin/B23, impairs its CK2-mediated phosphorylation, and leads to apoptosis through its nucleolar disassembly activity. Mol. Cancer Ther. 2009, 8, 1189–1196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobaina, Y.; Perera, Y. Implication of B23/NPM1 in viral infections, potential uses of B23/NPM1 inhibitors as antiviral therapy. Infect. Disord. Drug Targets 2019, 19, 2–16. [Google Scholar] [CrossRef]
- Shi, D.; Shi, H.; Sun, D.; Chen, J.; Zhang, X.; Wang, X.; Zhang, J.; Ji, Z.; Liu, J.; Cao, L.; et al. Nucleocapsid interacts with NPM1 and protects it from proteolytic cleavage, enhancing cell survival, and is involved in PEDV growth. Sci. Rep. 2017, 7, 39700. [Google Scholar] [CrossRef] [Green Version]
- Yip, S.P.; Siu, P.M.; Leung, P.H.; Zhao, Y.; Yung, B.Y. The multifunctional nucleolar protein nucleophosmin/NPM/B23 and the nucleoplasmin family of proteins. In The Nucleolus; Springer: New York, NY, USA, 2011; pp. 213–252. [Google Scholar]
- Miranda, J.; Bringas, R.; Fernandez-de-Cossio, J.; Perera-Negrin, Y. Targeting CK2 mediated signaling to impair/tackle SARS-CoV-2 infection: A computational biology approach. Mol. Med. Camb. Mass. 2021, 27, 161. [Google Scholar] [CrossRef]
- Kamel, W.; Noerenberg, M.; Cerikan, B.; Chen, H.; Järvelin, A.I.; Kammoun, M.; Lee, J.; Shuai, N.; Garcia-Moreno, M.; Andrejeva, A. Global analysis of protein-RNA interactions in SARS-CoV-2 infected cells reveals key regulators of infection. Mol. Cell 2021, 81, 2851–2867. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramón, A.C.; Pérez, G.V.; Caballero, E.; Rosales, M.; Aguilar, D.; Vázquez-Blomquist, D.; Ramos, Y.; Rodríguez-Ulloa, A.; Falcón, V.; Rodríguez-Moltó, M.P.; et al. Targeting of Protein Kinase CK2 Elicits Antiviral Activity on Bovine Coronavirus Infection. Viruses 2022, 14, 552. https://doi.org/10.3390/v14030552
Ramón AC, Pérez GV, Caballero E, Rosales M, Aguilar D, Vázquez-Blomquist D, Ramos Y, Rodríguez-Ulloa A, Falcón V, Rodríguez-Moltó MP, et al. Targeting of Protein Kinase CK2 Elicits Antiviral Activity on Bovine Coronavirus Infection. Viruses. 2022; 14(3):552. https://doi.org/10.3390/v14030552
Chicago/Turabian StyleRamón, Ailyn C., George V. Pérez, Evelin Caballero, Mauro Rosales, Daylén Aguilar, Dania Vázquez-Blomquist, Yassel Ramos, Arielis Rodríguez-Ulloa, Viviana Falcón, María Pilar Rodríguez-Moltó, and et al. 2022. "Targeting of Protein Kinase CK2 Elicits Antiviral Activity on Bovine Coronavirus Infection" Viruses 14, no. 3: 552. https://doi.org/10.3390/v14030552
APA StyleRamón, A. C., Pérez, G. V., Caballero, E., Rosales, M., Aguilar, D., Vázquez-Blomquist, D., Ramos, Y., Rodríguez-Ulloa, A., Falcón, V., Rodríguez-Moltó, M. P., Yang, K., Perera, Y., & Perea, S. E. (2022). Targeting of Protein Kinase CK2 Elicits Antiviral Activity on Bovine Coronavirus Infection. Viruses, 14(3), 552. https://doi.org/10.3390/v14030552