
Genomic Characterisation of UFJF_PfDIW6: A Novel Lytic Pseudomonas fluorescens-Phage with Potential for Biocontrol in the Dairy Industry

 ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Phage Isolation and Propagation
2.2. Phage DNA Extraction and Sequencing
2.3. Quality Control, Trimming and Filtering
2.4. Genome Assembly, Gene Prediction and Protein Annotation
2.5. Endolysin Screening
2.6. Comparative Genomics
2.7. Taxonomy Assignment and Phylogenetic Analysis
3. Results and Discussion
3.1. UFJF_PfDIW6’s Genome Features
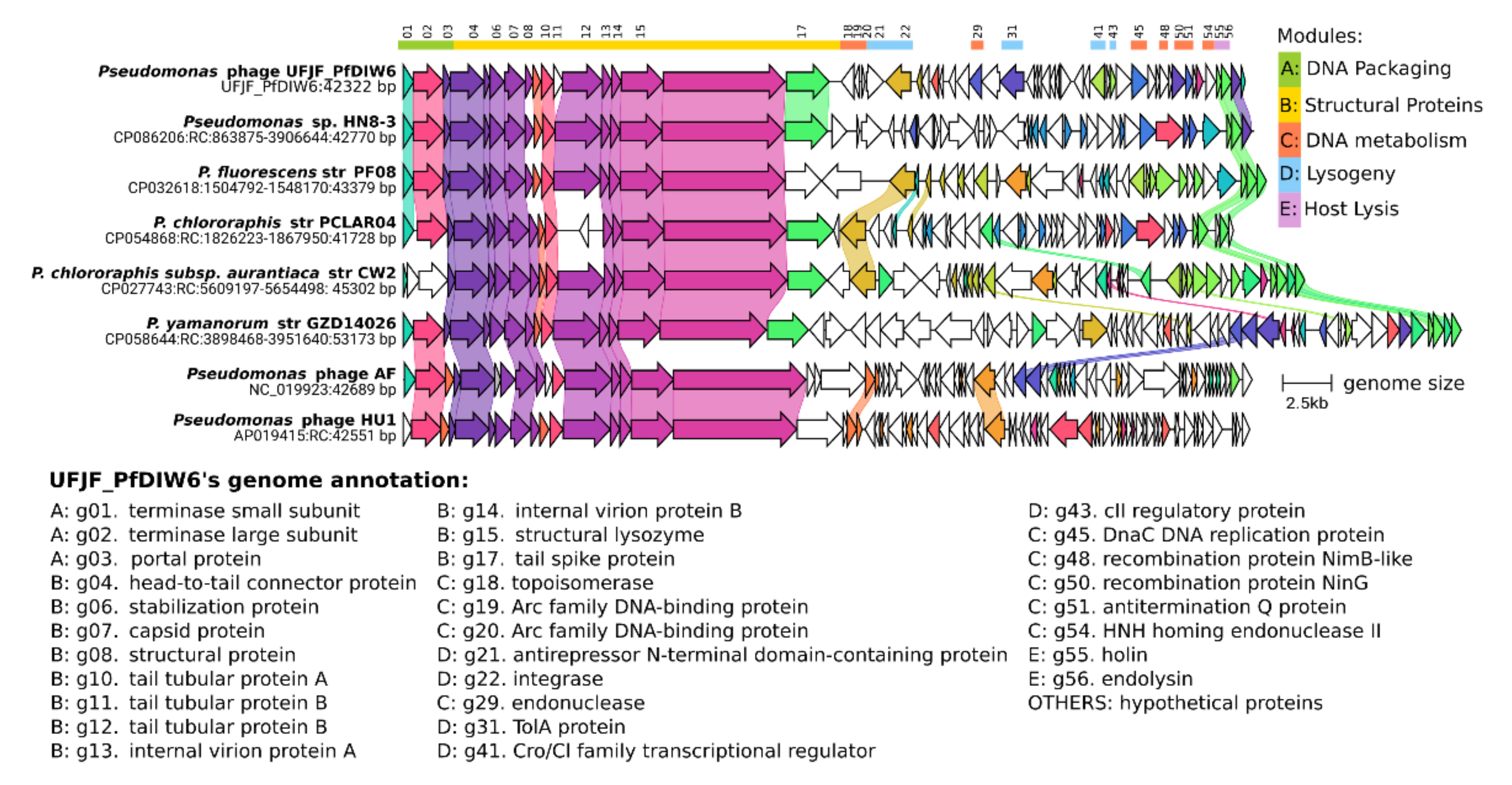
3.2. Comparative Genomics
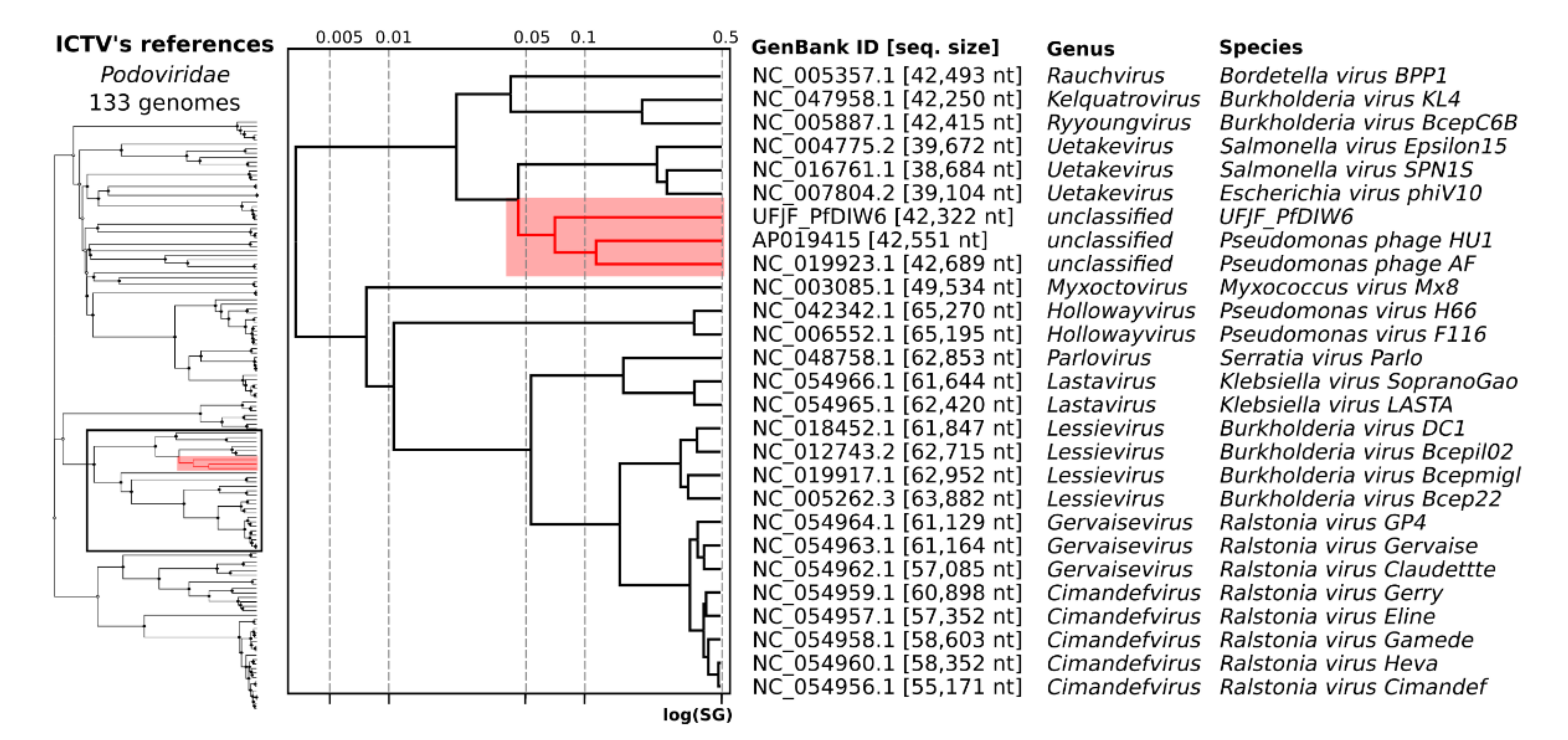
3.3. Taxonomic Assignment
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Smet, J.; Hendrix, H.; Blasdel, B.G.; Danis-Wlodarczyk, K.; Lavigne, R. Pseudomonas predators: Understanding and exploiting phage–host interactions. Nat. Rev. Microbiol. 2017, 15, 517–530. [Google Scholar] [CrossRef]
- Silby, M.W.; Winstanley, C.; Godfrey, S.A.C.; Levy, S.B.; Jackson, R.W. Pseudomonas Genomes: Diverse and adaptable. FEMS Microbiol. Rev. 2011, 35, 652–680. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, R.d.S.; Machado, S.G.; de Carvalho, A.F.; Nero, L.A. Pseudomonas sp. as the causative agent of anomalous blue discoloration in Brazilian fresh soft cheese (Minas Frescal). Int. Dairy J. 2021, 117, 105020. [Google Scholar] [CrossRef]
- Machado, S.G.; Baglinière, F.; Marchand, S.; Van Coillie, E.; Vanetti, M.C.D.; De Block, J.; Heyndrickx, M. The Biodiversity of the Microbiota Producing Heat-Resistant Enzymes Responsible for Spoilage in Processed Bovine Milk and Dairy Products. Front. Microbiol. 2017, 8, 302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, L.; Zhang, Y.; Liu, H.; Zhao, S.; Wang, J.; Zheng, N. Characterization of Pseudomonas spp. and Associated Proteolytic Properties in Raw Milk Stored at Low Temperatures. Front. Microbiol. 2017, 8, 2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribeiro Júnior, J.C.; Teider Junior, P.I.; Oliveira, A.L.M.; Rios, E.A.; Tamanini, R.; Beloti, V. Proteolytic and Lipolytic Potential of Pseudomonas spp. from Goat and Bovine Raw Milk. Pesq. Vet. Bras. 2018, 38, 1577–1583. [Google Scholar] [CrossRef]
- Matselis, E.; Roussis, I.G. Proteinase and lipase production by Pseudomonas fluorescens. Proteolysis and lipolysis in thermized ewe’s milk. Food Control 1998, 9, 251–259. [Google Scholar] [CrossRef]
- Endersen, L.; Coffey, A. The use of bacteriophages for food safety. Curr. Opin. Food Sci. 2020, 36, 1–8. [Google Scholar] [CrossRef]
- O’Sullivan, L.; Bolton, D.; Mcauliffe, O.; Coffey, A. Bacteriophages in Food Applications: From Foe to Friend. Annu. Rev. Food Sci. Technol. 2018, 10, 151–172. [Google Scholar] [CrossRef]
- O’Sullivan, L.; Bolton, D.; McAuliffe, O.; Coffey, A. The use of bacteriophages to control and detect pathogens in the dairy industry. Int. J. Dairy Technol. 2020, 73, 1–11. [Google Scholar] [CrossRef]
- García-Anaya, M.C.; Sepulveda, D.R.; Sáenz-Mendoza, A.I.; Rios-Velasco, C.; Zamudio-Flores, P.B.; Acosta-Muñiz, C.H. Phages as biocontrol agents in dairy products. Trends Food Sci. Technol. 2020, 95, 10–20. [Google Scholar] [CrossRef]
- Garcia, P.; Madera, C.; Martínez, B.; Rodriguez, A.; Suárez, J.E. Prevalence of bacteriophages infecting Staphylococcus aureus in dairy samples and their potential as biocontrol agents. J. Dairy Sci. 2009, 92, 3019–3026. [Google Scholar] [CrossRef] [PubMed]
- O’Flaherty, S.; Coffey, A.; Meaney, W.J.; Fitzgerald, G.F.; Ross, R.P. Inhibition of bacteriophage K proliferation on Staphylococcus aureus in raw bovine milk. Lett. Appl. Microbiol. 2005, 41, 274–279. [Google Scholar] [CrossRef] [PubMed]
- García, P.; Madera, C.; Martínez, B.; Rodríguez, A. Biocontrol of Staphylococcus aureus in curd manufacturing processes using bacteriophages. Int. Dairy J. 2007, 17, 1232–1239. [Google Scholar] [CrossRef]
- El Haddad, L.; Roy, J.-P.; Khalil, G.E.; St-Gelais, D.; Champagne, C.P.; Labrie, S.; Moineau, S. Efficacy of two Staphylococcus aureus phage cocktails in cheese production. Int. J. Food Microbiol. 2016, 217, 7–13. [Google Scholar] [CrossRef]
- Silva, E.N.G.; Figueiredo, A.C.L.; Miranda, F.A.; Almeida, R.C.d.C. Control of Listeria monocytogenes growth in soft cheeses by bacteriophage P100. Braz. J. Microbiol. 2014, 45, 11–16. [Google Scholar] [CrossRef] [Green Version]
- Perera, M.N.; Abuladze, T.; Li, M.; Woolston, J.; Sulakvelidze, A. Bacteriophage cocktail significantly reduces or eliminates Listeria monocytogenes contamination on lettuce, apples, cheese, smoked salmon and frozen foods. Food Microbiol. 2015, 52, 42–48. [Google Scholar] [CrossRef]
- Carlton, R.M.; Noordman, W.H.; Biswas, B.; de Meester, E.D.; Loessner, M.J. Bacteriophage P100 for control of Listeria monocytogenes in foods: Genome sequence, bioinformatic analyses, oral toxicity study, and application. Regul. Toxicol. Pharmacol. 2005, 43, 301–312. [Google Scholar] [CrossRef]
- Guenther, S.; Loessner, M.J. Bacteriophage biocontrol of Listeria monocytogenes on soft ripened white mold and red-smear cheeses. Bacteriophage 2011, 1, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Guenther, S.; Huwyler, D.; Richard, S.; Loessner, M.J. Virulent Bacteriophage for Efficient Biocontrol of Listeria monocytogenes in Ready-To-Eat Foods. Appl. Environ. Microbiol. 2009, 75, 93–100. [Google Scholar] [CrossRef] [Green Version]
- McLean, S.K.; Dunn, L.A.; Palombo, E.A. Phage Inhibition of Escherichia coli in Ultrahigh-Temperature-Treated and Raw Milk. Foodborne Pathog. Dis. 2013, 10, 956–962. [Google Scholar] [CrossRef] [PubMed]
- Grygorcewicz, B.; Chajęcka-Wierzchowska, W.; Augustyniak, A.; Wasak, A.; Stachurska, X.; Nawrotek, P.; Dołęgowska, B. In-milk inactivation of Escherichia coli O157:H7 by the environmental lytic bacteriophage ECPS-6. J. Food Saf. 2020, 40. [Google Scholar] [CrossRef]
- Modi, R.; Hirvi, Y.; Hill, A.; Griffiths, M.W. Effect of Phage on Survival of Salmonella Enteritidis during Manufacture and Storage of Cheddar Cheese Made from Raw and Pasteurized Milk. J. Food Prot. 2001, 64, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Zinno, P.; Devirgiliis, C.; Ercolini, D.; Ongeng, D.; Mauriello, G. Bacteriophage P22 to challenge Salmonella in foods. Int. J. Food Microbiol. 2014, 191, 69–74. [Google Scholar] [CrossRef]
- Tanaka, C.; Yamada, K.; Takeuchi, H.; Inokuchi, Y.; Kashiwagi, A.; Toba, T. A Lytic Bacteriophage for Controlling Pseudomonas lactis in Raw Cow’s Milk. Appl. Environ. Microbiol. 2018, 84, e00111-18. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Meng, X.-C.; Liu, F. Isolation and characterisation of lytic bacteriophages against Pseudomonas spp., a novel biological intervention for preventing spoilage of raw milk. Int. Dairy J. 2016, 55, 72–78. [Google Scholar] [CrossRef]
- do Nascimento, E.C.; Sabino, M.C.; Corguinha, L.d.R.; Targino, B.N.; Lange, C.C.; Pinto, C.L.d.O.; Pinto, P.d.F.; Vidigal, P.M.P.; Sant’Ana, A.S.; Hungaro, H.M. Lytic bacteriophages UFJF_PfDIW6 and UFJF_PfSW6 prevent Pseudomonas fluorescens growth in vitro and the proteolytic-caused spoilage of raw milk during chilled storage. Food Microbiol. 2022, 101, 103892. [Google Scholar] [CrossRef]
- Fernández, L.; Escobedo, S.; Gutiérrez, D.; Portilla, S.; Martínez, B.; García, P.; Rodríguez, A. Bacteriophages in the Dairy Environment: From Enemies to Allies. Antibiotics 2017, 6, 27. [Google Scholar] [CrossRef] [Green Version]
- Eller, M.R.; Salgado, R.L.; Vidigal, P.M.P.; Alves, M.P.; Dias, R.S.; de Oliveira, L.L.; da Silva, C.C.; de Carvalho, A.F.; de Paula, S.O. Complete Genome Sequence of the Pseudomonas fluorescens Bacteriophage UFV-P2. Genome Announc. 2013, 1, e00006-12. [Google Scholar] [CrossRef] [Green Version]
- Koberg, S.; Gieschler, S.; Brinks, E.; Wenning, M.; Neve, H.; Franz, C.M.A.P. Genome sequence of the novel virulent bacteriophage PMBT14 with lytic activity against Pseudomonas fluorescens DSM 50090R. Arch. Virol. 2018, 163, 2575–2577. [Google Scholar] [CrossRef]
- Sillankorva, S.; Neubauer, P.; Azeredo, J. Isolation and Characterization of a T7-like Lytic Phage for Pseudomonas fluorescens. BMC Biotechnol. 2008, 8, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.-J.; Liu, C.-F.; Yu, L.; Cui, F.-J.; Zhou, Q.; Yu, S.-L.; Sun, L. A novel bacteriophage KSL-1 of 2-Keto-gluconic acid producer Pseudomonas fluorescens K1005: Isolation, characterization and its remedial action. BMC Microbiol. 2012, 12, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wojtus, J.K.; Fitch, J.L.; Christian, E.; Dalefield, T.; Lawes, J.K.; Kumar, K.; Peebles, C.L.; Altermann, E.; Hendrickson, H.L. Complete Genome Sequences of Three Novel Pseudomonas fluorescens SBW25 Bacteriophages, Noxifer, Phabio, and Skulduggery. Genome Announc. 2017, 5, e00725-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, Y.; Wang, S.; Li, J.; Wei, Y.; Zhang, Q.; Lin, L.; Ji, X. Isolation and characterization of two lytic cold-active bacteriophages infecting Pseudomonas fluorescens from the Napahai plateau wetland. Can. J. Microbiol. 2018, 64, 183–190. [Google Scholar] [CrossRef]
- Zhang, C.; Zhang, Z.; Li, J.; Qin, K.; Wei, Y.; Zhang, Q.; Lin, L.; Xiuling, J. Complete genome sequence of the lytic cold-active Pseudomonas fluorescens bacteriophage VSW-3 from Napahai plateau wetland. Virus Genes 2016, 53, 146–150. [Google Scholar] [CrossRef]
- Turner, D.; Kropinski, A.M.; Adriaenssens, E.M. A Roadmap for Genome-Based Phage Taxonomy. Viruses 2021, 13, 506. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; p. 999. [Google Scholar]
- Summer, E.J. Preparation of a Phage DNA Fragment Library for Whole Genome Shotgun Sequencing. Methods Mol. Biol. 2009, 502, 27–46. [Google Scholar] [CrossRef]
- Krueger, F.; James, F.; Ewels, P.; Afyounian, E.; Schuster-Boeckler, B. FelixKrueger/TrimGalore: v0.6.7-DOI via Zenodo. 2021. Available online: https://zenodo.org/record/5127899#.YjMyx3rMJPZ (accessed on 26 October 2021).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Boetzer, M.; Henkel, C.V.; Jansen, H.J.; Butler, D.; Pirovano, W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 2011, 27, 578–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, M.; Rocha, M.; Oliveira, H.; Dias, O. Predicting promoters in phage genomes using PhagePromoter. Bioinformatics 2019, 35, 5301–5302. [Google Scholar] [CrossRef] [PubMed]
- Lesnik, E.A.; Sampath, R.; Levene, H.B.; Henderson, T.J.; McNeil, J.A.; Ecker, D.J. Prediction of rho-independent transcriptional terminators in Escherichia coli. Nucleic Acids Res. 2001, 29, 3583–3594. [Google Scholar] [CrossRef] [Green Version]
- Villarroel, J.; Kleinheinz, K.A.; Jurtz, V.I.; Zschach, H.; Lund, O.; Nielsen, M.; Larsen, M.V. HostPhinder: A Phage Host Prediction Tool. Viruses 2016, 8, 116. [Google Scholar] [CrossRef]
- Hockenberry, A.J.; Wilke, C.O. BACPHLIP: Predicting Bacteriophage Lifestyle from Conserved Protein Domains. PeerJ 2021, 9, e11396. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Eddy, S.R. Accelerated Profile HMM Searches. PLOS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Potter, S.C.; Luciani, A.; Eddy, S.R.; Park, Y.; López, R.; Finn, R.D. HMMER web server: 2018 update. Nucleic Acids Res. 2018, 46, W200–W204. [Google Scholar] [CrossRef] [Green Version]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gontijo, M.T.P.; Vidigal, P.M.P.; Lopez, M.E.S.; Brocchi, M. Bacteriophages that infect Gram-negative bacteria as source of signal-arrest-release motif lysins. Res. Microbiol. 2021, 172, 103794. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.; Melo, L.D.R.; Santos, S.B.; Nóbrega, F.L.; Ferreira, E.C.; Cerca, N.; Azeredo, J.; Kluskens, L.D. Molecular Aspects and Comparative Genomics of Bacteriophage Endolysins. J. Virol. 2013, 87, 4558–4570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almagro Armenteros, J.J.; Tsirigos, K.D.; Sønderby, C.K.; Petersen, T.N.; Winther, O.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 2019, 37, 420–423. [Google Scholar] [CrossRef] [PubMed]
- Hiller, K.; Grote, A.; Scheer, M.; Münch, R.; Jahn, D. PrediSi: Prediction of signal peptides and their cleavage positions. Nucleic Acids Res. 2004, 32, W375–W379. [Google Scholar] [CrossRef]
- Hirokawa, T.; Boon-Chieng, S.; Mitaku, S. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics 1998, 14, 378–379. [Google Scholar] [CrossRef] [Green Version]
- Käll, L.; Krogh, A.; Sonnhammer, E.L. A Combined Transmembrane Topology and Signal Peptide Prediction Method. J. Mol. Biol. 2004, 338, 1027–1036. [Google Scholar] [CrossRef]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The TOPCONS web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef]
- Darling, A.E.; Mau, B.; Perna, N.T.; Batzoglou, S.; Zhong, Y. progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss and Rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilchrist, C.L.M.; Chooi, Y.-H. clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Moraru, C.; Varsani, A.; Kropinski, A.M. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Li, M.; Zeng, Y.; Hu, X.; Tan, Y.; Rao, X.; Jin, X.; Li, S.; Zhu, J.; Zhang, K.; et al. Functional Identifica-tion of the DNA Packaging Terminase from Pseudomonas aeruginosa Phage PaP3. Arch. Virol. 2012, 157, 2133–2141. [Google Scholar] [CrossRef] [Green Version]
- Fujisawa, H.; Morita, M. Phage DNA packaging. Genes Cells 1997, 2, 537–545. [Google Scholar] [CrossRef]
- Prevelige, P.E.; Cortines, J.R. Phage assembly and the special role of the portal protein. Curr. Opin. Virol. 2018, 31, 66–73. [Google Scholar] [CrossRef]
- Leiman, P.G.; Shneider, M.M. Contractile Tail Machines of Bacteriophages. Adv. Exp. Med. Biol. 2012, 726, 93–114. [Google Scholar] [CrossRef] [PubMed]
- Hardies, S.C.; Thomas, J.A.; Black, L.; Weintraub, S.T.; Hwang, C.Y.; Cho, B.C. Identification of structural and morphogenesis genes of Pseudoalteromonas phage φRIO-1 and placement within the evolutionary history of Podoviridae. Virology 2016, 489, 116–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, B.; Margolin, W.; Molineux, I.J.; Liu, J. The Bacteriophage T7 Virion Undergoes Extensive Structural Re-modeling during Infection. Science 2013, 339, 576–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Champoux, J.J. DNA Topoisomerases: Structure, Function, and Mechanism. Annu. Rev. Biochem. 2003, 70, 369–413. [Google Scholar] [CrossRef] [Green Version]
- Learn, B.A.; Um, S.-J.; Huang, L.; McMacken, R. Cryptic single-stranded-DNA binding activities of the phage λ P and Escherichia coli DnaC replication initiation proteins facilitate the transfer of E. coli DnaB helicase onto DNA. Proc. Natl. Acad. Sci. USA 1997, 94, 1154–1159. [Google Scholar] [CrossRef] [Green Version]
- Quiles-Puchalt, N.; Carpena, N.; Alonso, J.C.; Novick, R.P.; Marina, A.; Penadés, J.R. Staphylococcal Pathogenicity Island DNA Packaging System Involving Cos-Site Packaging and Phage-Encoded HNH Endonucleases. Proc. Natl. Acad. Sci. USA 2014, 111, 6016–6021. [Google Scholar] [CrossRef] [Green Version]
- Kala, S.; Cumby, N.; Sadowski, P.D.; Hyder, B.Z.; Kanelis, V.; Davidson, A.R.; Maxwell, K.L. HNH Proteins Are a Widespread Component of Phage DNA Packaging Machines. Proc. Natl. Acad. Sci. USA 2014, 111, 6022–6027. [Google Scholar] [CrossRef] [Green Version]
- Hochhut, B.; Wilde, C.; Balling, G.; Middendorf, B.; Dobrindt, U.; Brzuszkiewicz, E.; Gottschalk, G.; Carniel, E.; Hacker, J. Role of pathogenicity island-associated integrases in the genome plasticity of uropathogenic Escherichia coli strain 536. Mol. Microbiol. 2006, 61, 584–595. [Google Scholar] [CrossRef]
- Wilde, C.; Mazel, D.; Hochhut, B.; Middendorf, B.; Le Roux, F.; Carniel, E.; Dobrindt, U.; Hacker, J. Delineation of the recombination sites necessary for integration of pathogenicity islands II and III into the Escherichia coli 536 chromosome. Mol. Microbiol. 2008, 68, 139–151. [Google Scholar] [CrossRef]
- Feiner, R.; Argov, T.; Rabinovich, L.; Sigal, N.; Borovok, I.; Herskovits, A.A. A new perspective on lysogeny: Prophages as active regulatory switches of bacteria. Nat. Rev. Microbiol. 2015, 13, 641–650. [Google Scholar] [CrossRef]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broussard, G.W.; Oldfield, L.M.; Villanueva, V.M.; Lunt, B.L.; Shine, E.E.; Hatfull, G.F. Integration-Dependent Bacteriophage Immunity Provides Insights into the Evolution of Genetic Switches. Mol. Cell 2013, 49, 237–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, Y.; Shin, H.; Lee, J.-H.; Park, C.J.; Paik, S.-Y.; Ryu, S. Isolation and Genome Characterization of the Virulent Staphylococcus aureus Bacteriophage SA97. Viruses 2015, 7, 5225–5242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, Q.; Trinh, J.T.; Zeng, L. High-resolution studies of lysis–lysogeny decision-making in bacteriophage lambda. J. Biol. Chem. 2019, 294, 3343–3349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boeckaerts, D.; Stock, M.; Criel, B.; Gerstmans, H.; De Baets, B.; Briers, Y. Predicting bacteriophage hosts based on sequences of annotated receptor-binding proteins. Sci. Rep. 2021, 11, 1467. [Google Scholar] [CrossRef]
- Valero-Rello, A. Diversity, specificity and molecular evolution of the lytic arsenal of Pseudomonas phages: In silico perspective. Environ. Microbiol. 2019, 21, 4136–4150. [Google Scholar] [CrossRef]
- Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef]
- Wang, I.-N.; Smith, D.L.; Young, R. Holins: The Protein Clocks of Bacteriophage Infections. Annu. Rev. Microbiol. 2000, 54, 799–825. [Google Scholar] [CrossRef]
- Catalao, M.J.; Gil, F.; Moniz-Pereira, J.; São-José, C.; Pimentel, M. Diversity in bacterial lysis systems: Bacteriophages show the way. FEMS Microbiol. Rev. 2013, 37, 554–571. [Google Scholar] [CrossRef] [Green Version]
- Rajaure, M.; Berry, J.; Kongari, R.; Cahill, J.; Young, R. Membrane Fusion during Phage Lysis. Proc. Natl. Acad. Sci. USA 2015, 112, 5497–5502. [Google Scholar] [CrossRef] [Green Version]
- Crispim, J.S.; Dias, R.S.; Vidigal, P.M.P.; De Sousa, M.P.; Da Silva, C.C.; Santana, M.F.; De Paula, S.O. Screening and characterization of prophages in Desulfovibrio genomes. Sci. Rep. 2018, 8, 9273. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, A.; Ceyssens, P.-J.; Krylov, V.N.; Noben, J.-P.; Volckaert, G.; Lavigne, R. Identification of EPS-degrading activity within the tail spikes of the novel Pseudomonas putida phage AF. Virology 2012, 434, 251–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hungaro, H.M.; Vidigal, P.M.P.; do Nascimento, E.C.; Gomes da Costa Oliveira, F.; Gontijo, M.T.P.; Lopez, M.E.S. Genomic Characterisation of UFJF_PfDIW6: A Novel Lytic Pseudomonas fluorescens-Phage with Potential for Biocontrol in the Dairy Industry. Viruses 2022, 14, 629. https://doi.org/10.3390/v14030629
Hungaro HM, Vidigal PMP, do Nascimento EC, Gomes da Costa Oliveira F, Gontijo MTP, Lopez MES. Genomic Characterisation of UFJF_PfDIW6: A Novel Lytic Pseudomonas fluorescens-Phage with Potential for Biocontrol in the Dairy Industry. Viruses. 2022; 14(3):629. https://doi.org/10.3390/v14030629
Chicago/Turabian StyleHungaro, Humberto Moreira, Pedro Marcus Pereira Vidigal, Edilane Cristina do Nascimento, Felipe Gomes da Costa Oliveira, Marco Túlio Pardini Gontijo, and Maryoris Elisa Soto Lopez. 2022. "Genomic Characterisation of UFJF_PfDIW6: A Novel Lytic Pseudomonas fluorescens-Phage with Potential for Biocontrol in the Dairy Industry" Viruses 14, no. 3: 629. https://doi.org/10.3390/v14030629
APA StyleHungaro, H. M., Vidigal, P. M. P., do Nascimento, E. C., Gomes da Costa Oliveira, F., Gontijo, M. T. P., & Lopez, M. E. S. (2022). Genomic Characterisation of UFJF_PfDIW6: A Novel Lytic Pseudomonas fluorescens-Phage with Potential for Biocontrol in the Dairy Industry. Viruses, 14(3), 629. https://doi.org/10.3390/v14030629