
Genetic Analysis of the Complete S1 Gene in Japanese Infectious Bronchitis Virus Strains


Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses
2.2. RNA Extraction, RT-PCR, and Sequencing
2.3. Genetic Analysis of the S1 Gene
2.4. Phylogenetic Analysis
2.5. Recombination Analysis
3. Results
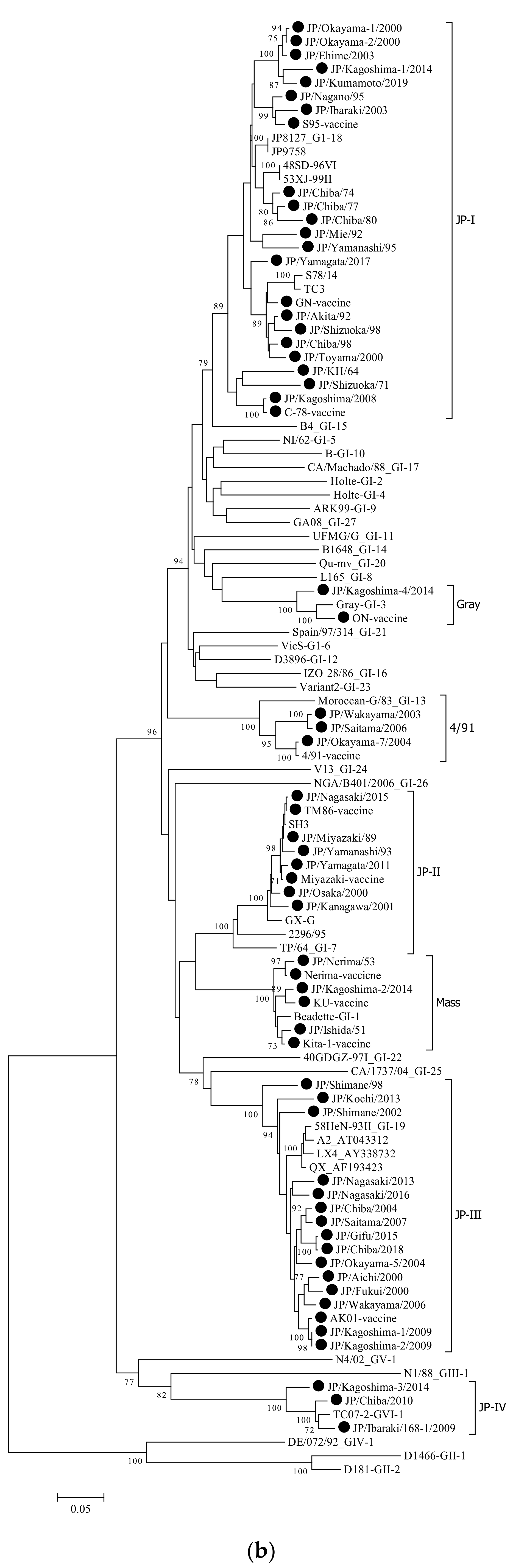
3.1. Phylogenetic Analysis
3.2. Recombination Analysis
3.3. Proteolytic Cleavage Sites in the Spike Glycoprotein
3.4. Amino Acids Related to the Receptor-Binding Domain (RBD)
3.5. Potential N-glycosylation Sites
4. Discussion
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Jackwood, M.; de Wit, J.J. Infectious bronchitis virus. In Diseases of Poultry, 14th ed.; Swayne, D.E., Boulianne, M., Logue, C.M., McDougald, L.R., Nair, V., Suarez, D.L., Eds.; Wiley-Blackwell: Hoboken, NJ, USA, 2020; pp. 167–188. [Google Scholar]
- De Wit, J.J.; Cook, J.K.A. Spotlight on avian pathology: Infectious bronchitis virus. Avian Pathol. 2019, 48, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B. Vaccination against infectious bronchitis virus: A continuous challenge. Vet. Microbiol. 2017, 206, 137–143. [Google Scholar] [CrossRef]
- Cavanagh, D. Coronavirus avian infectious bronchitis virus. Vet. Res. 2007, 38, 281–297. [Google Scholar] [CrossRef] [PubMed]
- Koch, G.; Hartog, L.; Kant, A.; Van Roozelaar, D.J. Antigenic domains on the peplomer protein of avian infectious bronchitis virus: Correlation with biological functions. J. Gen. Virol. 1990, 71, 1929–1935. [Google Scholar] [CrossRef]
- Kant, A.; Koch, G.; Van Roozelaar, D.J.; Kusters, J.G.; Poelwijk, F.A.J.; Van der Zeijst, B.A.M. Location of antigenic sites defined by neutralizing monoclonal antibodies on the S1 avian infectious bronchitis virus glycopolypeptide. J. Gen. Virol. 1992, 73, 591–596. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.K.; Jeon, W.J.; Lee, Y.J.; Jeong, O.M.; Choi, J.G.; Kwon, J.H.; Choi, K.S. Genetic diversity of avian infectious bronchitis virus isolates in Korea between 2003 and 2006. Avian Dis. 2008, 52, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Zhang, L.; Hou, Y.; Zhao, Y.; Han, Z.; Sun, J.; Liu, S. Genetic, antigenic, and pathogenic characteristics of infectious bronchitis virus GI-7/TW-II in China. Avian Dis. 2020, 64, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.H.; Huang, Y.C. Relationship between serotypes and genotypes based on the hypervariable region of the S1 gene of infectious bronchitis virus. Arch. Virol. 2000, 145, 291–300. [Google Scholar] [CrossRef]
- Valastro, V.; Holmes, E.C.; Britton, P.; Fusaro, A.; Jackwood, M.W.; Cattoli, G.; Monne, I. S1 gene-based phylogeny of infectious bronchitis virus: An attempt to harmonize virus classification. Infect. Genet. Evol. 2016, 39, 349–364.11. [Google Scholar] [CrossRef]
- Mase, M.; Tsukamoto, K.; Imai, K.; Yamaguchi, S. Phylogenetic analysis of avian infectious bronchitis virus strains isolated in Japan. Arch. Virol. 2004, 149, 2069–2078. [Google Scholar] [CrossRef]
- Mase, M.; Gotou, M.; Inoue, D.; Watanabe, S.; Iseki, H. Genotyping of infectious bronchitis viruses isolated in Japan during 2008−2019. J. Vet. Med Sci. 2021, 83, 522–526. [Google Scholar] [CrossRef] [PubMed]
- Shang, J.; Zheng, Y.; Yang, Y.; Liu, C.; Geng, Q.; Luo, C.; Zhang, W.; Li, F. Cryo-EM structure of infectious bronchitis coronavirus spike protein reveals structural and functional evolution of coronavirus spike proteins. PLoS Pathog. 2018, 14. [Google Scholar] [CrossRef] [PubMed]
- Parsons, L.M.; Bouwman, K.M.; Azurmendi, H.; De Vries, R.P.; Cipollo, J.F.; Verheije, M.H. Glycosylation of the viral attachment protein of avian coronavirus is essential for host cell and receptor binding. J. Biol. Chem. 2019, 294, 7797–7809. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Lim, T.H.; Lee, H.J.; Lee, D.H.; Lee, Y.N.; Park, J.K.; Youn, H.N.; Kim, M.S.; Lee, J.B.; Park, S.Y.; Choi, I.S.; et al. An emerging recombinant cluster of nephropathogenic strains of avian infectious bronchitis virus in Korea. Infect. Genet. Evol. 2011, 11, 678–685. [Google Scholar] [CrossRef]
- Pohuang, T.; Chansiripornchai, N.; Tawatsin, A.; Sasipreeyajan, J. Sequence analysis of S1 genes of infectious bronchitis virus isolated in Thailand during 2008-2009: Identification of natural recombination in the field isolates. Virus Genes 2011, 43, 254–260. [Google Scholar] [CrossRef]
- Promkuntod, N.; van Eijndhoven, R.E.W.; de Vrieze, G.; Gröne, A.; Verheije, M.H. Mapping of the receptor-binding domain and amino acids critical for attachment in the spike protein of avian coronavirus infectious bronchitis virus. Virology 2014, 448, 26–32. [Google Scholar] [CrossRef]
- Sun, X.; Li, L.; Pan, L.; Wang, Z.; Chen, H.; Shao, C.; Yu, J.; Ren, Y.; Wang, X.; Huang, X.; et al. Infectious bronchitis virus: Identification of Gallus gallus APN high-affinity ligands with antiviral effects. Antivir. Res. 2021, 186. [Google Scholar] [CrossRef]
- Bouwman, K.M.; Parsons, L.M.; Berends, A.J.; de Vries, R.P.; Cipollo, J.F.; Verheije, M.H. Three Amino Acid Changes in Avian Coronavirus Spike Protein Allow Binding to Kidney Tissue. J. Virol. 2020, 94, e01363-19. [Google Scholar] [CrossRef]
- Zheng, J.; Yamada, Y.; Fung, T.S.; Huang, M.; Chia, R.; Liu, D.X. Identification of N-linked glycosylation sites in the spike protein and their functional impact on the replication and infectivity of coronavirus infectious bronchitis virus in cell culture. Virology 2018, 513, 65–74. [Google Scholar] [CrossRef]
- Stevenson-Leggett, P.; Armstrong, S.; Keep, S.; Britton, P.; Bickerton, E. Analysis of the avian coronavirus spike protein reveals heterogeneity in the glycans present. J. Gen. Virol. 2021, 102. [Google Scholar] [CrossRef] [PubMed]
- Wickramasinghe, I.N.A.; de Vries, R.P.; Gröne, A.; de Haan, C.A.M.; Verheije, M.H. Binding of Avian Coronavirus Spike Proteins to Host Factors Reflects Virus Tropism and Pathogenicity. J. Virol. 2011, 85, 8903–8912. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Cheng, X.; Zhao, X.; Yu, Y.; Gao, M.; Zhou, S. Recombinant infectious bronchitis coronavirus H120 with the spike protein S1 gene of the nephropathogenic IBYZ strain remains attenuated but induces protective immunity. Vaccine 2020, 38, 3157–3168. [Google Scholar] [CrossRef] [PubMed]
- Jackwood, M.W.; Hilt, D.A.; Callison, S.A.; Lee, C.W.; Plaza, H.; Wade, E. Spike glycoprotein cleavage recognition site analysis of infectious bronchitis virus. Avian Dis. 2001, 45, 366–372. [Google Scholar] [CrossRef]
- Zhang, X.; Deng, T.; Lu, J.; Zhao, P.; Chen, L.; Qian, M.; Guo, Y.; Qiao, H.; Xu, Y.; Wang, Y.; et al. Molecular characterization of variant infectious bronchitis virus in China, 2019: Implications for control programmes. Transbound. Emerg. Dis. 2020, 67, 1349–1355. [Google Scholar] [CrossRef]
- Abro, S.H.; Ullman, K.; Belák, S.; Baule, C. Bioinformatics and evolutionary insight on the spike glycoprotein gene of QX-like and Massachusetts strains of infectious bronchitis virus. Virol. J. 2012, 9. [Google Scholar] [CrossRef]
- Casais, R.; Thiel, V.; Siddell, S.G.; Cavanagh, D.; Britton, P. Reverse genetics system for the avian coronavirus infectious bronchitis virus. J. Virol. 2001, 75, 12359–12369. [Google Scholar] [CrossRef]
- Van Beurden, S.J.; Berends, A.J.; Krämer-Kühl, A.; Spekreijse, D.; Chénard, G.; Philipp, H.C.; Mundt, E.; Rottier, P.J.M.; Verheije, M.H. A reverse genetics system for avian coronavirus infectious bronchitis virus based on targeted RNA recombination. Virol. J. 2017, 14. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Strain | Isolation Year | Length (bp) | Clinical Signs | Genotype Based on HVR-1,2 | Genotype Based on Complete Sequence of S1 Gene | Cleavage Site |
|---|---|---|---|---|---|---|
| JP/KH/64 | 1964 | 1632 | Respiratory | JP-I | GI-18 | RRSRR |
| JP/Shizuoka/71 | 1971 | 1632 | Respiratory | JP-I | GI-18 | RRSRR |
| JP/Chiba/74 | 1974 | 1632 | Respiratory | JP-I | GI-18 | RRSRR |
| JP/Chiba/77 | 1977 | 1632 | Egg drop | JP-I | GI-18 | RRSRR |
| JP/Chiba/80 | 1980 | 1632 | Respiratory | JP-I | GI-18 | RRSRR |
| JP/Mie/92 | 1992 | 1629 | Nephritis | JP-I | GI-18 | HRFRR |
| JP/Akita/92 | 1992 | 1632 | Respiratory | JP-I | GI-18 | RRFKR |
| JP/Nagano/95 | 1995 | 1629 | Nephritis | JP-I | GI-18 | RRSKR |
| JP/Yamanashi/95 | 1995 | 1632 | Respiratory | JP-I | GI-18 | RRSRR |
| JP/Shizuoka/98 | 1998 | 1626 | Nephritis | JP-I | GI-18 | RRSRR |
| JP/Chiba/98 | 1998 | 1632 | Nephritis | JP-I | GI-18 | RRFKR |
| JP/Toyama/2000 | 2000 | 1632 | Nephritis | JP-I | GI-18 | HRFRR |
| JP/Okayama-1/2000 | 2000 | 1632 | Nephritis | JP-I | GI-18 | RRFKR |
| JP/Okayama-2/2000 | 2000 | 1632 | Nephritis | JP-I | GI-18 | RRFKR |
| JP/Ibaraki/2003 | 2003 | 1632 | Nephritis | JP-I | GI-18 | RRSKR |
| JP/Ehime/2003 | 2003 | 1632 | Depression, respiratory | JP-I | GI-18 | RRFKR |
| JP/Kagoshima/2008 | 2008 | 1629 | Depression, respiratory | JP-I | GI-18 | RRSRR |
| JP/Kagoshima-1/2014 | 2014 | 1632 | Nephritis | JP-I | GI-18 | RRFRR |
| JP/Yamagata/2017 | 2017 | 1629 | Rise in mortality | JP-I | GI-18 | RRSRR |
| JP/Kumamoto/2019 | 2019 | 1632 | Nephritis | JP-I | GI-18 | RRFRR |
| C-78 | 1629 | Vaccine strain | JP-I | GI-18 | RRSRR | |
| GN | 1632 | Vaccine strain | JP-I | GI-18 | RRFKR | |
| S95 | 1632 | Vaccine strain | JP-I | GI-18 | RRSKR | |
| JP/Miyazaki/89 | 1989 | 1614 | Nephritis | JP-II | GI-7 | RRFRR |
| JP/Yamanashi/93 | 1993 | 1614 | Nephritis | JP-II | GI-7 | RRFRR |
| JP/Osaka/2000 | 2000 | 1614 | Nephritis | JP-II | GI-7 | RRFRR |
| JP/Kanagawa/2001 | 2001 | 1614 | Nephritis | JP-II | GI-7 | RRSKR |
| JP/Yamagata/2011 | 2011 | 1614 | Nephritis | JP-II | GI-7 | RRFKR |
| JP/Nagasaki/2015 | 2015 | 1614 | Respiratory | JP-II | GI-7 | RRFRR |
| Miyazaki | 1614 | Vaccine strain | JP-II | GI-7 | RRFRR | |
| TM86 | 1614 | Vaccine strain | JP-II | GI-7 | RRFRR | |
| JP/Shimane/98 | 1998 | 1617 | Respiratory | JP-III | GI-19 | HRFRR |
| JP/Aichi/2000 | 2000 | 1620 | Nephritis | JP-III | GI-19 | RRFRR |
| JP/Fukui/2000 | 2000 | 1620 | Respiratory | JP-III | GI-19 | RRFRR |
| JP/Shimane/2002 | 2002 | 1614 | Nephritis | JP-III | GI-19 | RRFRR |
| JP/Okayama-5/2004 | 2004 | 1611 | Egg drop | JP-III | GI-19 | RRFRR |
| JP/Chiba/2004 | 2004 | 1617 | Nephritis | JP-III | GI-19 | RRFRR |
| JP/Wakayama-13/2006 | 2006 | 1620 | Rise in mortality | JP-III | GI-19 | RRFRR |
| JP/Saitama/2007 | 2007 | 1617 | Egg drop | JP-III | GI-19 | RRFRR |
| JP/Kagoshima-1/2009 | 2009 | 1620 | Depression, diarrhea | JP-III | GI-19 | RRFRR |
| JP/Kagoshima-2/2009 | 2009 | 1620 | Egg drop | JP-III | GI-19 | RRFRR |
| JP/Kochi/2013 | 2013 | 1617 | Rise in mortality | JP-III | Recombinant | RRFRR |
| JP/Nagasaki/2013 | 2013 | 1620 | Depression, diarrhea | JP-III | Recombinant | RRFRR |
| JP/Gifu/2015 | 2015 | 1620 | Egg drop | JP-III | GI-19 | RRFRR |
| JP/Nagasaki/2016 | 2016 | 1617 | Respiratory, diarrhea | JP-III | Recombinant | RRFKR |
| JP/Chiba/2018 | 2018 | 1620 | Respiratory | JP-III | GI-19 | RRFRR |
| AK01 | 1620 | Vaccine strain | JP-III | GI-19 | RRFRR | |
| JP/Ibaraki/168-1/2009 | 2009 | 1638 | Egg drop | JP-IV | GVI-1 | HRRKR |
| JP/Chiba/2010 | 2010 | 1638 | Nephritis | JP-IV | GVI-1 | HRRKR |
| JP/Kagoshima-3/2014 | 2014 | 1638 | Respiratory | JP-IV | GVI-1 | HRRKR |
| JP/Ishida/51 | 1951 | 1611 | Respiratory | Mass | GI-1 | RRFRR |
| JP/Nerima/53 | 1953 | 1611 | Respiratory | Mass | GI-1 | RRFRR |
| JP/Kagoshima-2/2014 | 2014 | 1611 | Nephritis | Mass | GI-1 | RRFRR |
| Nerima | 1611 | Vaccine strain | Mass | GI-1 | RRFRR | |
| Kita-1 | 1605 | Vaccine strain | Mass | GI-1 | RRFRR | |
| KU | 1611 | Vaccine strain | Mass | GI-1 | RRFRR | |
| JP/Kagoshima-4/2014 | 2014 | 1623 | Rise in mortality | Gray | GI-3 | RRSRR |
| ON | 1629 | Vaccine strain | Gray | GI-3 | RRSRR | |
| JP/Wakayama/2003 | 2003 | 1617 | Depression, diarrhea | 4/91 | GI-13 | RRFRR |
| JP/Okayama-7/2004 | 2004 | 1617 | Nephritis | 4/91 | GI-13 | RRSRR |
| JP/Saitama/2006 | 2006 | 1617 | Respiratory | 4/91 | GI-13 | RRFRR |
| Name | Sequence (5ʹ-3ʹ) | Position a | Length (bp) | Reference | |
|---|---|---|---|---|---|
| Forward | Reverse | ||||
| 15F | AGGAATGGTAAGTTRCTRGTWAGAG | 20343–20367 | 671 | Mase et al., 2004 [11] | |
| 26Rm | GCGCAGTACCRTTRAYAAAATAAGC | 21013–20989 | Mase et al., 2004 [11] | ||
| 19F | GCAGTGTTTGTTACGCATTG | 20689–20708 | 1333 | in this study | |
| 1R | CATAACTAACATAAGGGCAA | 22021–22002 | in this study |
| Genotype | Amino Acid Position of Amino Acids at the S1 Glycoprotein | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 26 | 28 | 29 | 34 | 36 | 37 | 38 | 40 | 42 | 43 | 63 | 69 | |
| Mass (GI-1) | Y | Y | Y | F | P | P | D/N | W | L | H/Q | P/S | T/I |
| Gray (GI-3) | Y | Y | Y | F | P | P | N | W | L | H | S | A |
| JP-I GI-18) | Y | Y | Y | F/L/Y | P | P/S/G | L/F/P/S/V | W | L/V/I | H | S/H/A | A |
| JP-II (GI-7) | Y | Y | Y | F | P | P | D/N | W | L | Q | P/L/R | S |
| JP-III (GI-19) | Y | Y | Y | F | P | P/S | D/N/T/E | W | L | Q | P/S/N/T/A/Q | V |
| JP-IV (GVI-1) | Y | Y | Y | F | P | P | L/S | W | L | H/Y | G/H | A |
| 4/91 (GI-13) | Y | Y | Y | F | P | G | P/Q | W | L | H/Y | P/S | T |
| Genotype | Corresponding Position of Amino Acids at S1 Glycoprotein | ||
|---|---|---|---|
| 110 | 111 | 112 | |
| Ck/SWE/0658946/10 (QX) | K | I | P |
| Mass (GI-1) | M/V | L/V/I | Q |
| Gray (GI-3) | F/I | L | P |
| JP-I (GI-18) | L/F/R/S | I | Q/N/A/E |
| JP-II (GI-7) | F | V | P |
| JP-III (GI-19) | M/Q/L | I | P/K |
| JP-IV (GVI-1) | K/I | L | D/K/G |
| 4/91 (GI-13) | M | I | P |
| Strain | Genotype | Amino Acid Position at POTENTIAL N-glycosylation a | Number of Glycosylation Site | |||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| M41(AY851295) | Mass (GI-1) | 51 | - | 77 | - | 103 b | - | - | 144 | 163 | 178 | - | - | 212 | 237 | 247 | 264 | 271 | 276 | - | 306 | - | 425 | 447 | - | 513 | 530 | 17 |
| JP/Ishida/51(LC662594) | Mass (GI-1) | 51 | - | 77 | - | 103 | - | - | 144 | 163 | 178 | - | - | 212 | 237 | 247 | 264 | 271 | 276 | - | 306 | - | 425 | 447 | - | 513 | 530 | 17 |
| JP/Nerima/53(LC662595) | Mass (GI-1) | 51 | - | - | - | 103 | - | - | 144 | 163 | 178 | - | - | 212 | 237 | 247 | 264 | - | 276 | - | 306 | - | 425 | 447 | - | 513 | 530 | 15 |
| Gray(L14069) | GI-3 | 51 | - | 75 | - | 103 | - | - | 150 | 169 | 184 | - | - | 218 | 243 | 253 | 270 | 277 | 282 | - | 312 | - | 431 | 453 | - | 519 | 536 | 17 |
| JP/Kagoshima-4/2014(LC662600) | Gray (GI-3) | - | - | 75 | - | 103 | - | - | 148 | 167 | 182 | - | - | 216 | 241 | 251 | 268 | 275 | 280 | - | 310 | - | 429 | 451 | - | 517 | 534 | 16 |
| QX(MN548289) | QX (GI-19) | 52 | 55 | 76 | 92 | 104 | 117 | 141 | 147 | 166 | 181 | - | 200 | 215 | 240 | 250 | 267 | 274 | 279 | 282 | 309 | 405 | 428 | 450 | 457 | 516 | 533 | 21 |
| JP/Shimane/98(LC662575) | JP-III (GI-19) | 51 | 54 | - | - | 103 | - | 140 | 146 | 165 | 180 | - | - | 214 | 239 | 249 | 266 | 273 | 278 | 281 | 308 | - | 427 | 449 | - | 515 | 532 | 19 |
| JP/Aichi/2000(LC662576) | JP-III (GI-19) | 52 | 55 | - | - | 104 | - | 141 | 147 | 166 | 181 | - | - | 215 | 240 | 250 | 267 | 274 | 279 | 282 | 309 | - | 428 | 450 | - | 516 | - | 18 |
| JP/Fukui/2000(LC662577) | JP-III (GI-19) | 52 | 55 | 76 | - | 104 | - | 141 | 147 | 166 | 181 | - | - | 215 | 240 | 250 | 267 | 274 | 279 | - | 309 | 405 | 428 | 450 | - | 516 | - | 19 |
| JP/Shimane/2002(LC662578) | JP-III (GI-19) | 50 | 53 | 74 | - | 102 | - | 139 | 145 | 164 | 179 | - | - | 213 | 238 | 248 | 265 | 272 | 277 | 280 | 307 | - | 426 | 448 | - | 514 | - | 19 |
| JP/Kagoshima-1/2009(LC662583) | JP-III (GI-19) | 52 | - | 76 | - | 104 | - | 141 | 147 | 166 | 181 | - | - | 215 | 240 | 250 | 267 | 274 | 279 | 282 | 309 | - | 428 | 450 | - | 516 | - | 18 |
| 4/91(KF377577) | - | 54 | 75 | - | 103 | - | - | 146 | 165 | 180 | - | - | 214 | 239 | 249 | 266 | 273 | 278 | 281 | 308 | - | 427 | 449 | 456 | 515 | 533 | 19 | |
| JP/Wakayama/2003(LC662602) | 4/91 (GI-13) | - | 54 | 75 | - | 103 | - | - | 146 | 165 | 180 | - | - | 214 | 239 | 249 | 266 | 273 | 278 | - | 308 | - | 427 | 449 | 456 | 515 | 532 | 18 |
| JP/Okayama-7/2004(LC662603) | 4/91 (GI-13) | - | 54 | 75 | - | 103 | - | - | 146 | 165 | 180 | 184 | - | 214 | 239 | 249 | 266 | 273 | 278 | 281 | 308 | - | 427 | 449 | 456 | 515 | 532 | 20 |
| JP8127(AY296744) | GI-18 | 51 | - | 75 | - | 103 | - | - | 151 | 170 | 185 | - | - | 219 | 244 | 254 | 271 | 278 | 283 | - | 314 | - | 433 | 455 | - | 521 | 538 | 17 |
| JP/KH/64(LC634083) | JP-I (GI-18) | 51 | - | 75 | - | 103 | - | - | 151 | 170 | 185 | - | - | 219 | 244 | 254 | 271 | 278 | 283 | - | 313 | - | 432 | 454 | - | 520 | - | 16 |
| JP/Akita/92(LC662550) | JP-I (GI-18) | 51 | - | 75 | - | 103 | - | - | 151 | 170 | 185 | - | - | 219 | 244 | 254 | 271 | 278 | 283 | - | 313 | - | 432 | 454 | - | 520 | 537 | 17 |
| JP/Nagano/95(LC662551) | JP-I (GI-18) | 51 | - | 75 | 91 | 103 | - | - | 151 | 170 | 185 | - | - | 219 | 244 | 254 | 271 | 278 | 283 | - | 313 | - | 431 | 453 | - | 519 | 536 | 18 |
| JP/Kagoshima-1/2014(LC662561) | JP-I (GI-18) | 51 | - | 75 | - | 103 | - | - | 151 | 170 | 185 | - | - | 219 | 244 | 254 | 271 | 278 | 283 | - | 313 | - | 432 | 454 | 461 | 520 | 537 | 18 |
| TP/64(AY606320) | GI-7 | 51 | - | 77 | - | 103 | 116 | 139 | 145 | 164 | 179 | - | - | 213 | 238 | 248 | 265 | 272 | 277 | - | 308 | - | 427 | 449 | - | 515 | 532 | 19 |
| JP/Miyazaki/89(LC662567) | JP-II (GI-7) | 51 | - | 77 | - | 103 | 116 | 139 | 145 | 164 | 179 | - | - | 213 | 238 | 248 | 265 | 272 | 277 | 280 | 307 | - | 426 | 448 | - | 514 | 531 | 20 |
| JP/Nagasaki/2015(LC662572) | JP-II (GI-7) | 51 | - | - | - | 103 | 116 | 139 | 145 | 164 | 179 | - | - | 213 | 238 | 248 | 265 | 272 | 277 | 280 | 307 | - | 426 | 448 | - | 514 | 531 | 19 |
| TC07-2(GQ265948) | GVI-1 | - | 55 | 75 | - | 104 | - | - | 148 | 167 | 182 | - | - | 216 | 241 | 256 | - | 275 | 280 | - | 314 | 411 | - | 456 | - | 522 | 539 | 16 |
| JP/Ibaraki/168-1/2009(LC662591) | JP-IV (GVI-1) | - | 55 | 75 | - | 104 | - | - | 148 | 167 | 182 | - | - | 216 | 241 | 256 | - | 275 | 280 | - | 314 | - | - | 456 | - | 522 | 539 | 15 |
| JP/Kagoshima-3/2014(LC662593) | JP-IV (GVI-1) | - | 55 | 75 | - | 104 | - | - | 148 | 167 | - | - | - | 216 | 241 | 256 | - | 275 | 280 | - | 314 | - | - | 456 | - | 522 | 539 | 14 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mase, M.; Hiramatsu, K.; Watanabe, S.; Iseki, H. Genetic Analysis of the Complete S1 Gene in Japanese Infectious Bronchitis Virus Strains. Viruses 2022, 14, 716. https://doi.org/10.3390/v14040716
Mase M, Hiramatsu K, Watanabe S, Iseki H. Genetic Analysis of the Complete S1 Gene in Japanese Infectious Bronchitis Virus Strains. Viruses. 2022; 14(4):716. https://doi.org/10.3390/v14040716
Chicago/Turabian StyleMase, Masaji, Kanae Hiramatsu, Satoko Watanabe, and Hiroshi Iseki. 2022. "Genetic Analysis of the Complete S1 Gene in Japanese Infectious Bronchitis Virus Strains" Viruses 14, no. 4: 716. https://doi.org/10.3390/v14040716
APA StyleMase, M., Hiramatsu, K., Watanabe, S., & Iseki, H. (2022). Genetic Analysis of the Complete S1 Gene in Japanese Infectious Bronchitis Virus Strains. Viruses, 14(4), 716. https://doi.org/10.3390/v14040716