
Activation of the Ca2+/NFAT Pathway by Assembly of Hepatitis C Virus Core Protein into Nucleocapsid-like Particles


Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cells, Cell Culture and Viral Genomes
2.3. Analysis of Gene Expression
2.4. Preparation of Cell Lysate, SDS-PAGE and Western Blotting
2.5. Treatment with MG132
2.6. Indirect Immunofluorescence
2.7. Centrifugation Analysis of HCV Nucleocapsid-like Particels
2.8. Electron Microscopy
3. Results
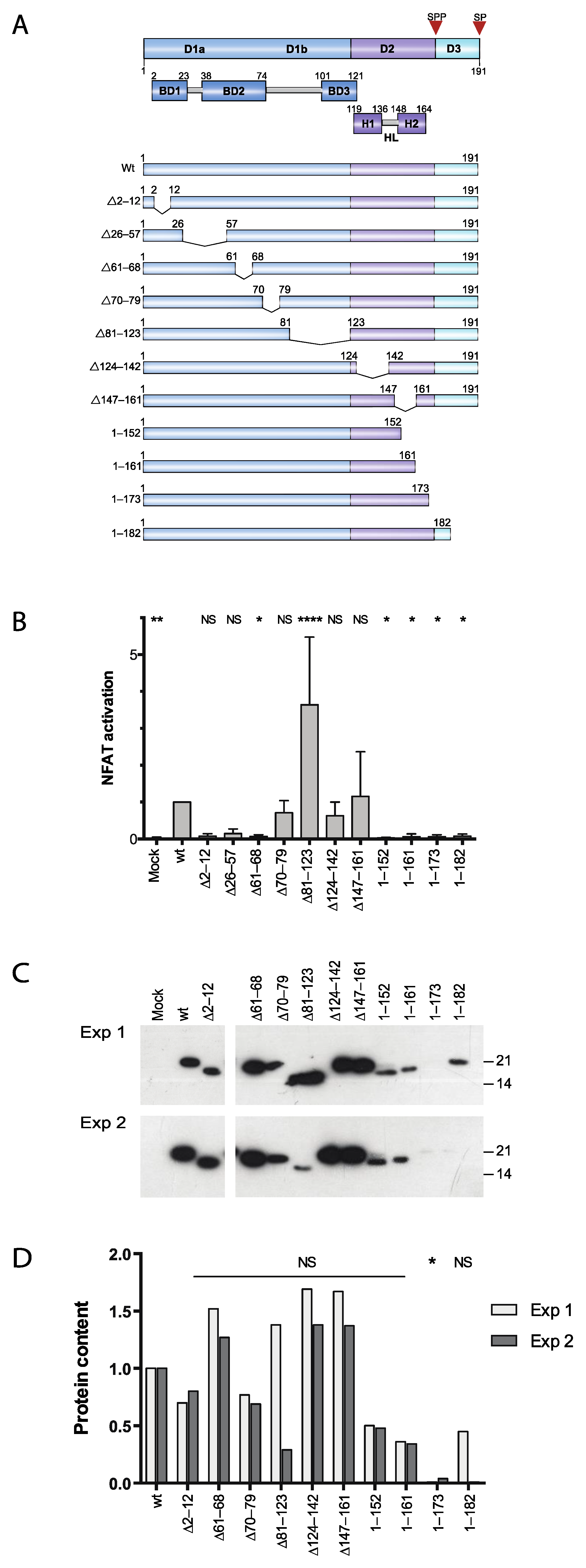
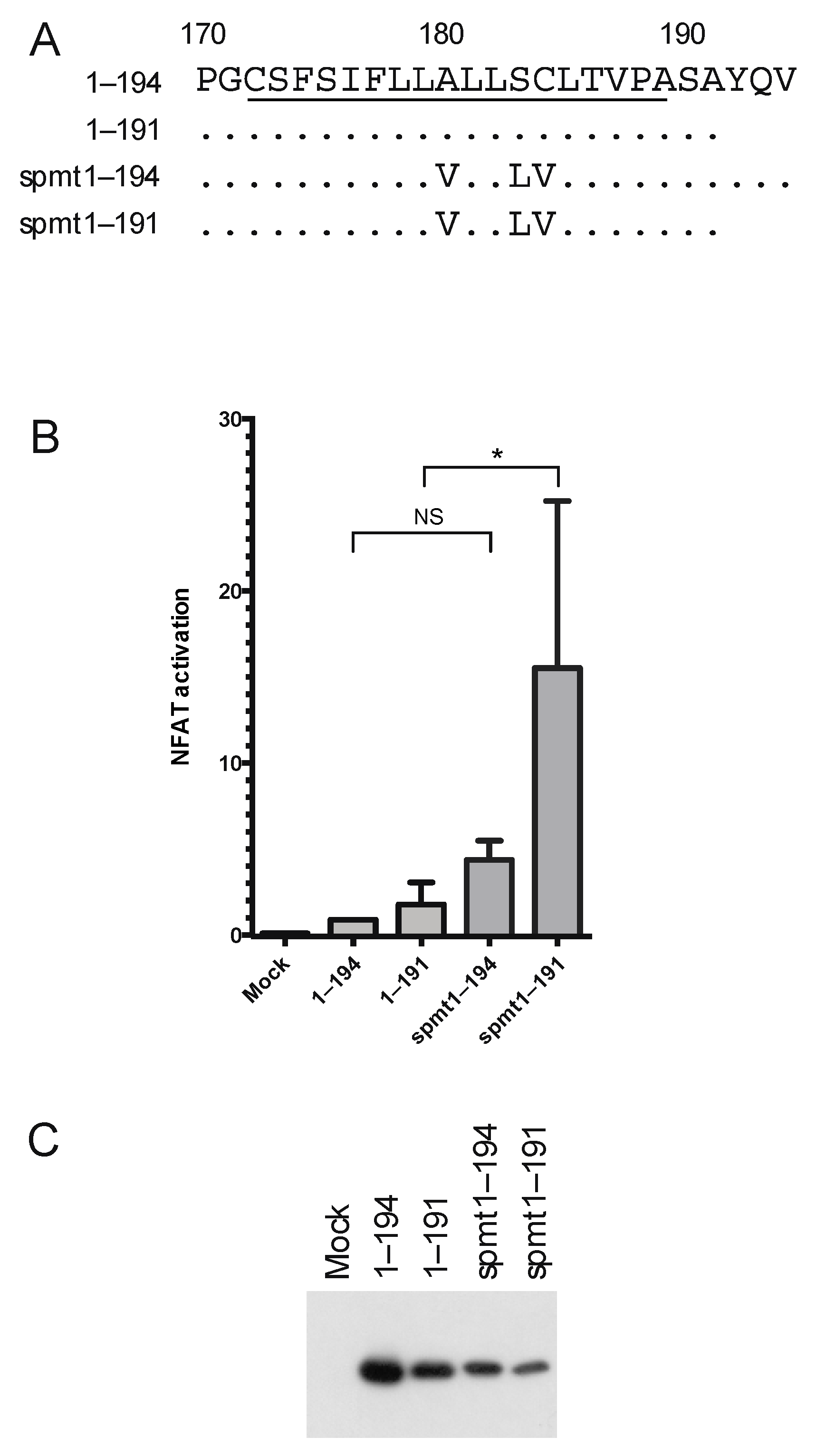
3.1. Two Distinct Regions of C Are Required for Activation of Ca2+/NFAT-Signaling
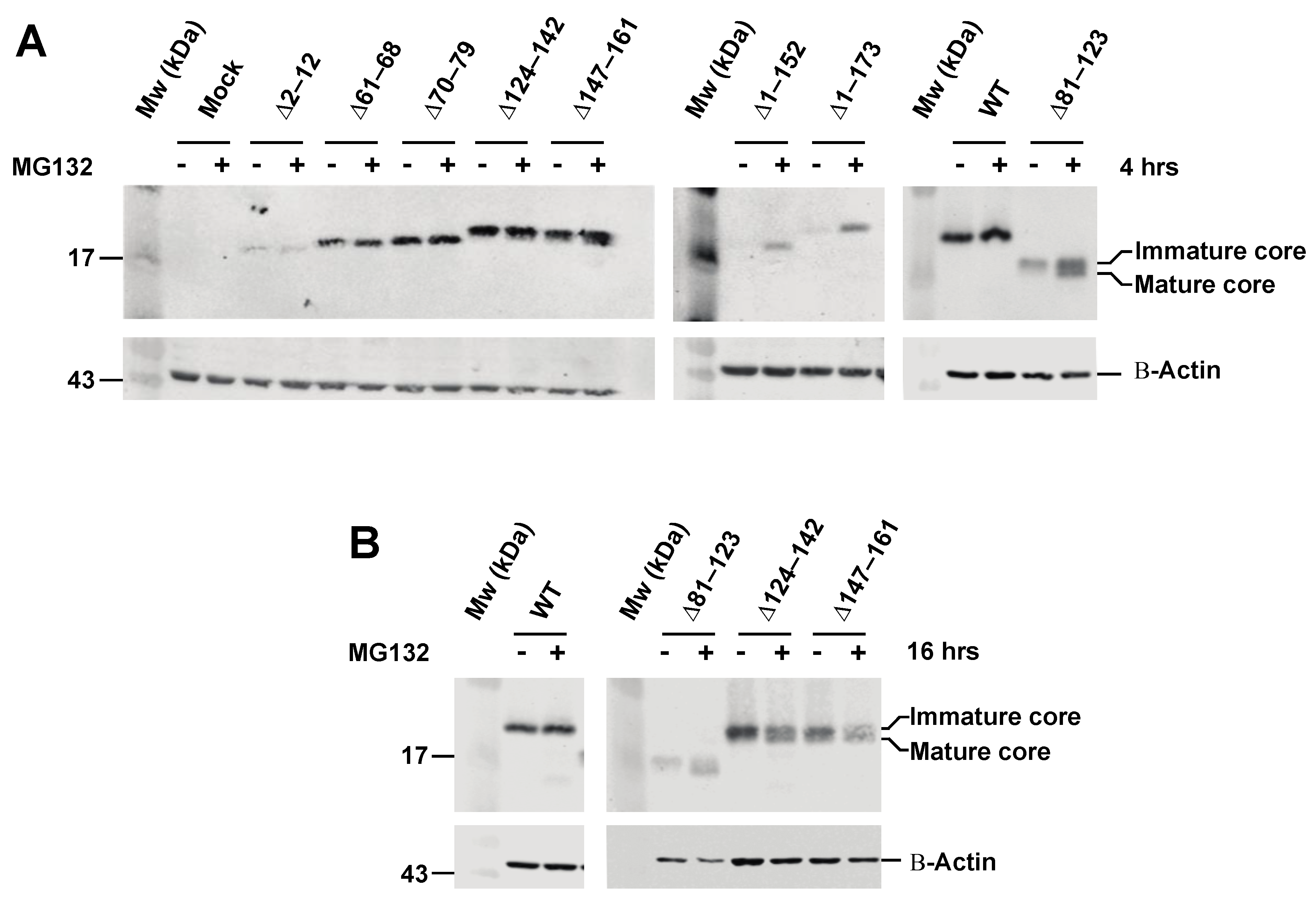
3.2. Effect of Proteasome Inhibiton on Expression of HCV C Proteins
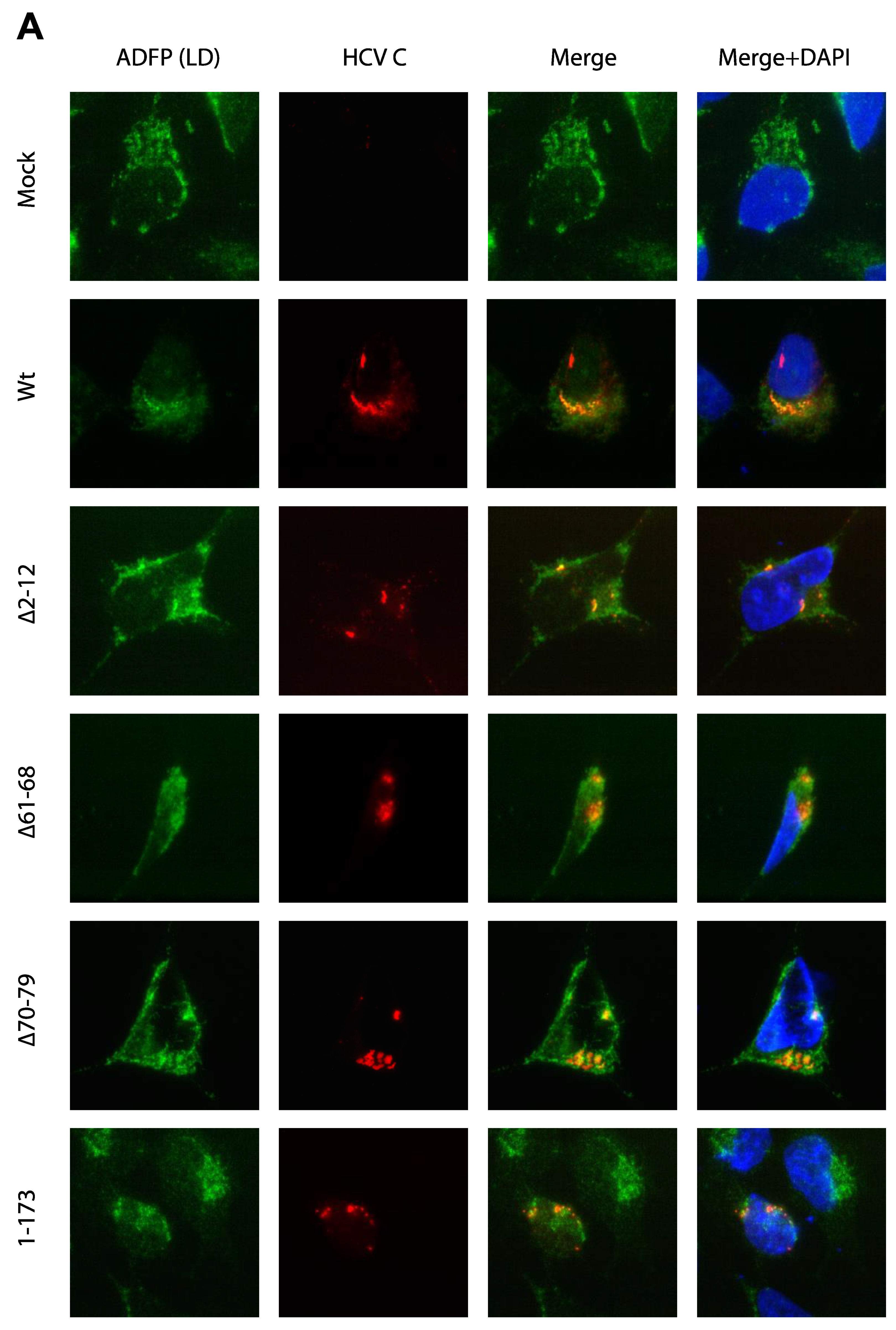
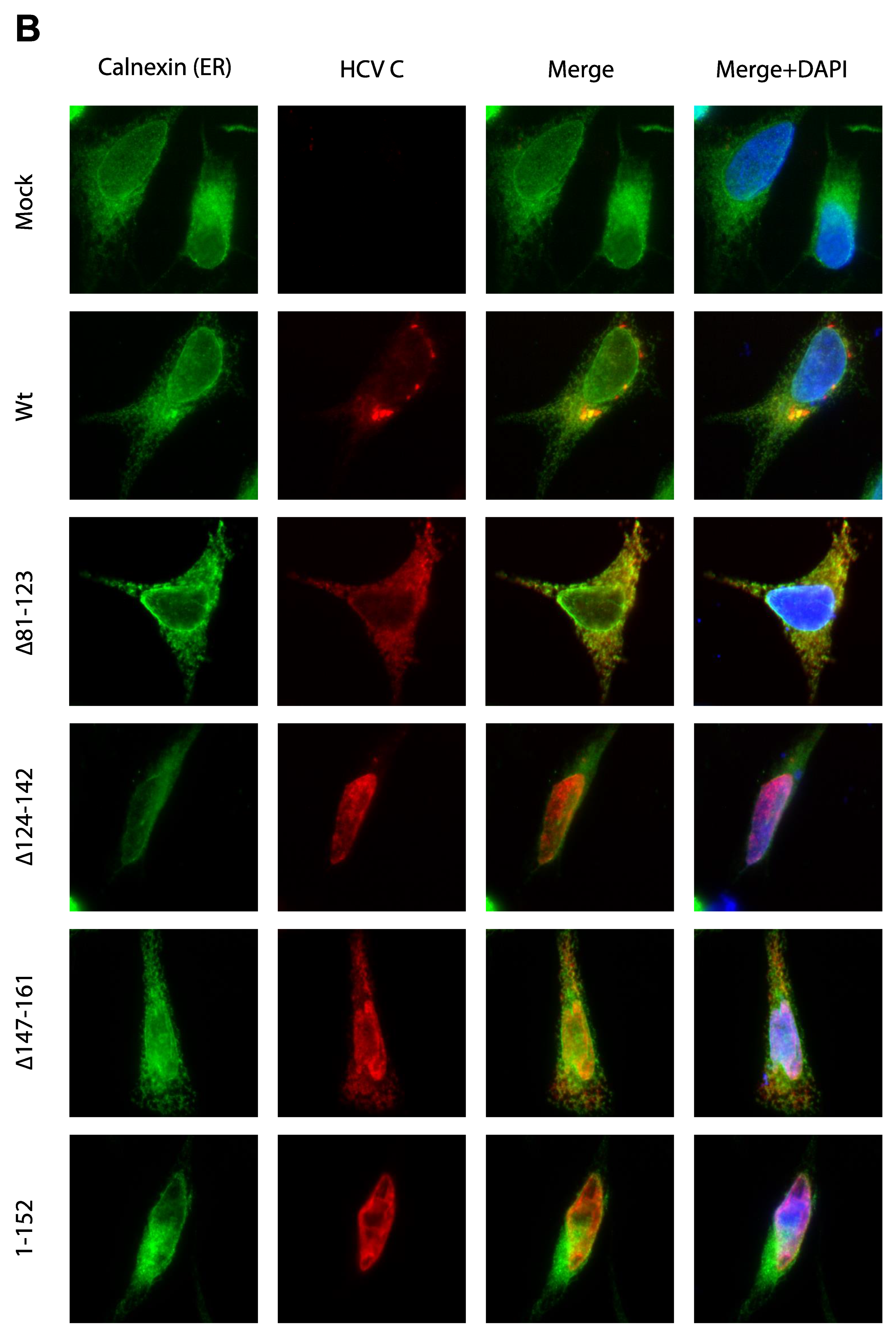
3.3. Activation of Ca2+/NFAT Signaling Is Unrelated to the C Protein Subcellular Localization
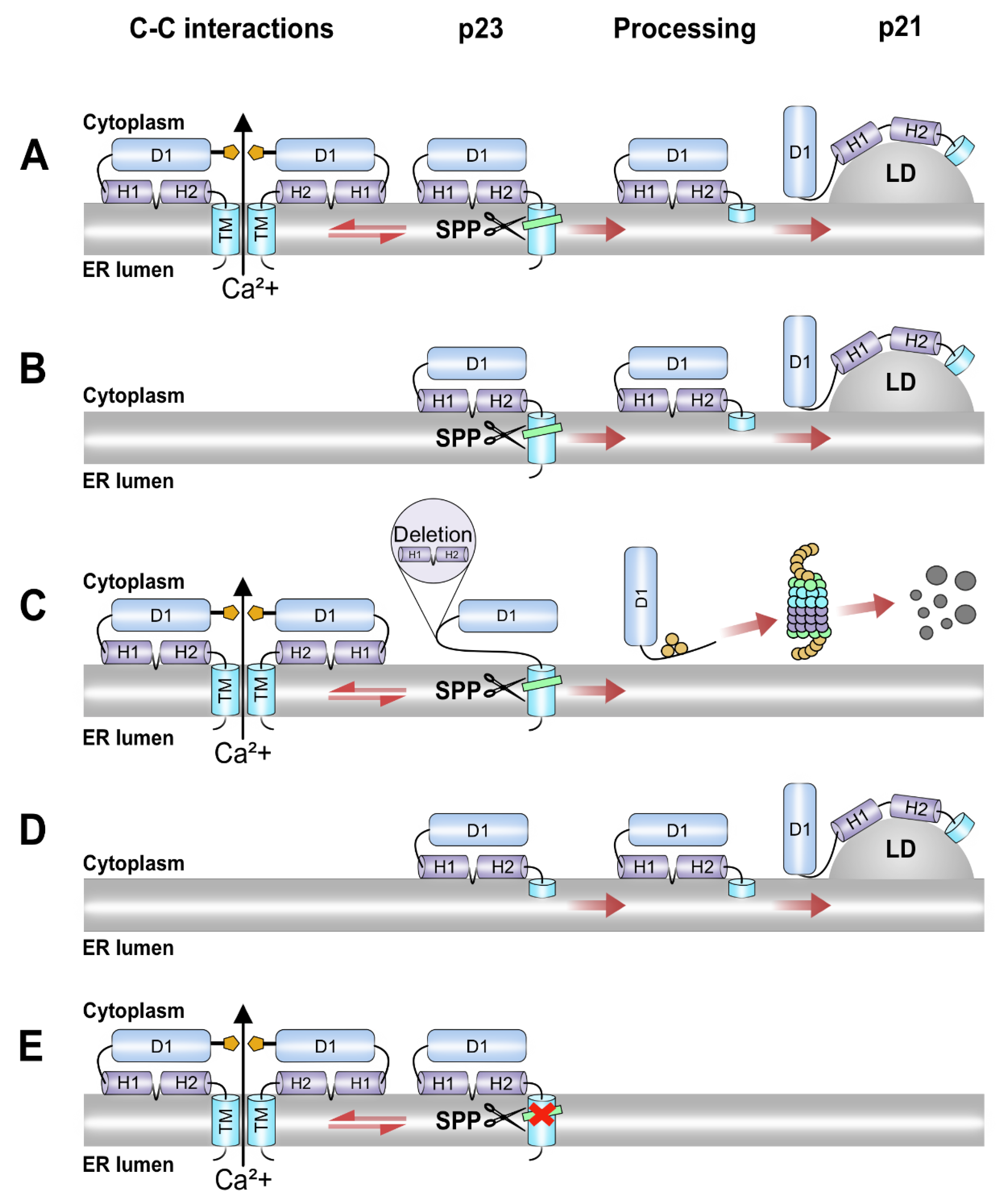
3.4. The Intermediate p23 Form of HCV C Is Responsible for Activation of Ca2+/NFAT-Signaling
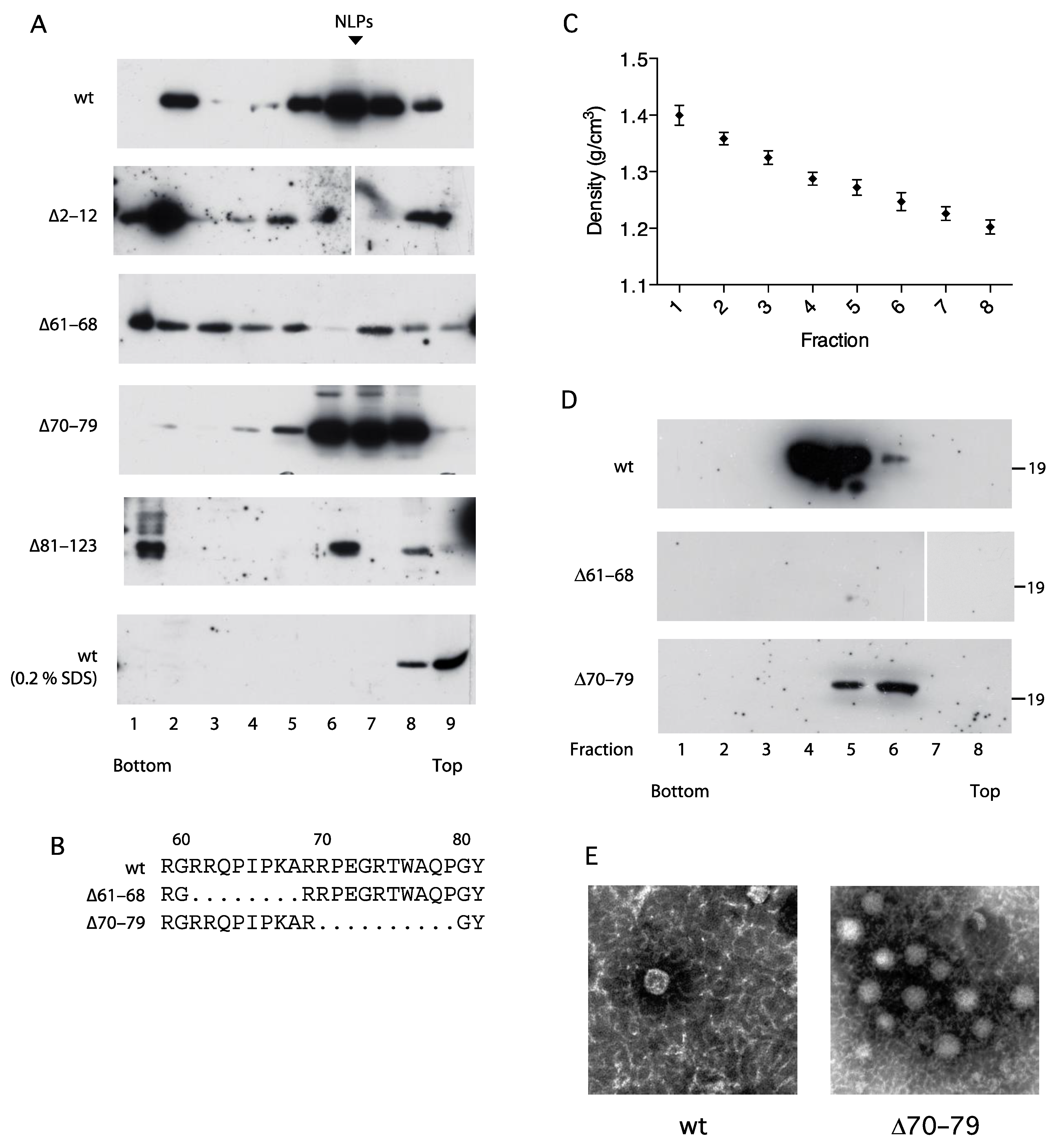
3.5. Association of the C Protein into Nucleocapsid-like Particles Correlates with Activation of the NFAT Signaling
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Spearman, C.W.; Dusheiko, G.M.; Hellard, M.; Sonderup, M. Hepatitis C. Lancet 2019, 394, 1451–1466. [Google Scholar] [CrossRef]
- Manns, M.P.; Buti, M.; Gane, E.; Pawlotsky, J.M.; Razavi, H.; Terrault, N.; Younossi, Z. Hepatitis C virus infection. Nat. Rev. Dis. Primers 2017, 3, 17006. [Google Scholar] [CrossRef] [PubMed]
- Reed, K.E.; Rice, C.M. Molecular Characterization of Hepatitis C Virus. Curr. Stud. Hematol. Blood Transfus. 1998, 62, 1–37. [Google Scholar] [CrossRef]
- Lindenbach, B.D.; Rice, C.M. Unravelling hepatitis C virus replication from genome to function. Nature 2005, 436, 933–938. [Google Scholar] [CrossRef] [PubMed]
- Mclauchlan, J.; Lemberg, M.K.; Hope, G.; Martoglio, B. Intramembrane proteolysis promotes trafficking of hepatitis C virus core protein to lipid droplets. EMBO J. 2002, 21, 3980–3988. [Google Scholar] [CrossRef] [PubMed]
- Mclauchlan, J. Properties of the hepatitis C virus core protein: A structural protein that modulates cellular processes. J. Viral Hepat. 2000, 7, 2–14. [Google Scholar] [CrossRef]
- Gawlik, K.; Gallay, P.A. HCV core protein and virus assembly: What we know without structures. Immunol. Res. 2014, 60, 1–10. [Google Scholar] [CrossRef]
- Tellinghuisen, T.L.; Rice, C.M. Interaction between hepatitis C virus proteins and host cell factors. Curr. Opin. Microbiol. 2002, 5, 419–427. [Google Scholar] [CrossRef]
- Ray, R.B.; Ray, R. Hepatitis C virus core protein: Intriguing properties and functional relevance. FEMS Microbiol. Lett. 2001, 202, 149–156. [Google Scholar] [CrossRef]
- Matsumori, A.; Matoba, Y.; Nishio, R.; Shioi, T.; Ono, K.; Sasayama, S. Detection of Hepatitis C Virus RNA from the Heart of Patients with Hypertrophic Cardiomyopathy. Biochem. Biophys. Res. Commun. 1996, 222, 678–682. [Google Scholar] [CrossRef]
- Laskus, T.; Radkowski, M.; Wang, L.-F.; Vargas, H.; Rakela, J. The Presence of Active Hepatitis C Virus Replication in Lymphoid Tissue in Patients Coinfected with Human Immunodeficiency Virus Type 1. J. Infect. Dis. 1998, 178, 1189–1192. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Sullivan, D.G.; Kim, S.; Lai, K.K.; Kae, J.; Cotler, S.J.; Carithers, R.L., Jr.; Wood, B.L.; Perkins, J.D.; Gretch, D.R. Productive Replication of Hepatitis C Virus in Perihepatic Lymph Nodes In Vivo: Implications of HCV Lymphotropism. Gastroenterology 2006, 130, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Blackard, J.T.; Kemmer, N.; Sherman, K.E. Extrahepatic replication of HCV: Insights into clinical manifestations and biological consequences. Hepatology 2006, 44, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Castillo, I.; Rodríguez-Iñigo, E.; Bartolomé, J.; de Lucas, S.; Ortíz-Movilla, N.; López-Alcorocho, J.M.; Pardo, M.; Carreño, V. Hepatitis C virus replicates in peripheral blood mononuclear cells of patients with occult hepatitis C virus infection. Gut 2005, 54, 682–685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, Y.; Machida, K.; Liu, H.M.; Ueno, Y.; Kobayashi, K.; Wakita, T.; Shimosegawa, T.; Lai, M.M.C. Hepatitis C Virus Infection of T Cells Inhibits Proliferation and Enhances Fas-Mediated Apoptosis by Down-Regulating the Expression of CD44 Splicing Variant 6. J. Infect. Dis. 2009, 199, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Ueno, Y.; Kakazu, E.; Kobayashi, K.; Shiina, M.; Tamai, K.; Machida, K.; Inoue, J.; Wakui, Y.; Fukushima, K.; et al. Lymphotropic HCV strain can infect human primary naïve CD4+ cells and affect their proliferation and IFN-γ secretion activity. J. Gastroenterol. 2011, 46, 232–241. [Google Scholar] [CrossRef] [Green Version]
- Kondo, Y.; Sung, V.M.; Machida, K.; Liu, M.; Lai, M.M. Hepatitis C virus infects T cells and affects interferon-gamma signaling in T cell lines. Virology 2007, 361, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Ray, R.B.; Ray, R. Oncogenic potential of hepatitis C virus proteins. Viruses 2010, 2, 2108–2133. [Google Scholar] [CrossRef]
- Moriya, K.; Fujie, H.; Shintani, Y.; Yotsuyanagi, H.; Tsutsumi, T.; Ishibashi, K.; Matsuura, Y.; Kimura, S.; Miyamura, T.; Koike, K. The core protein of hepatitis C virus induces hepatocellular carcinoma in transgenic mice. Nat. Med. 1998, 4, 1065–1067. [Google Scholar] [CrossRef]
- Moriya, K.; Yotsuyanagi, H.; Shintani, Y.; Fujie, H.; Ishibashi, K.; Matsuura, Y.; Miyamura, T.; Koike, K. Hepatitis C virus core protein induces hepatic steatosis in transgenic mice. J. Gen. Virol. 1997, 78 Pt 7, 1527–1531. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.-Y.; Chen, S.S.-L. Hepatitis C Virus and Cellular Stress Response: Implications to Molecular Pathogenesis of Liver Diseases. Viruses 2012, 4, 2251–2290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, S.W. Unfolded protein response in hepatitis C virus infection. Front. Microbiol. 2014, 5, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Green, R.M. Endoplasmic reticulum stress and liver diseases. Liver Res. 2019, 3, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and Oxidative Stress in the Liver. Viruses 2013, 5, 439–469. [Google Scholar] [CrossRef] [Green Version]
- Merquiol, E.; Uzi, D.; Mueller, T.; Goldenberg, D.; Nahmias, Y.; Xavier, R.J.; Tirosh, B.; Shibolet, O. HCV Causes Chronic Endoplasmic Reticulum Stress Leading to Adaptation and Interference with the Unfolded Protein Response. PLoS ONE 2011, 6, e24660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benali-Furet, N.L.; Chami, M.; Houel, L.; De Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; et al. Hepatitis C virus core triggers apoptosis in liver cells by inducing ER stress and ER calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS ONE 2011, 6, e24957. [Google Scholar] [CrossRef] [Green Version]
- Pal, S.; Polyak, S.J.; Bano, N.; Qiu, W.C.; Carithers, R.L.; Shuhart, M.; Gretch, D.R.; Das, A. Hepatitis C virus induces oxidative stress, DNA damage and modulates the DNA repair enzyme NEIL1. J. Gastroenterol. Hepatol. 2010, 25, 627–634. [Google Scholar] [CrossRef] [Green Version]
- Ríos-Ocampo, W.A.; Daemen, T.; Buist-Homan, M.; Faber, K.N.; Navas, M.C.; Moshage, H. Hepatitis C virus core or NS3/4A protein expression preconditions hepatocytes against oxidative stress and endoplasmic reticulum stress. Redox Rep. 2019, 24, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Ke, P.-Y.; Chen, S.S.-L. Activation of the unfolded protein response and autophagy after hepatitis C virus infection suppresses innate antiviral immunity in vitro. J. Clin. Investig. 2011, 121, 37–56. [Google Scholar] [CrossRef] [Green Version]
- Dionisio, N.; Garcia-Mediavilla, M.V.; Sanchez-Campos, S.; Majano, P.L.; Benedicto, I.; Rosado, J.A.; Salido, G.M.; Gonzalez-Gallego, J. Hepatitis C virus NS5A and core proteins induce oxidative stress-mediated calcium signalling alterations in hepatocytes. J. Hepatol. 2009, 50, 872–882. [Google Scholar] [CrossRef] [PubMed]
- Bergqvist, A.; Sundström, S.; Dimberg, L.Y.; Gylfe, E.; Masucci, M.G. The Hepatitis C Virus Core Protein Modulates T Cell Responses by Inducing Spontaneous and Altering T-Cell Receptor-Triggered Ca2+ Oscillations. J. Biol. Chem. 2003, 278, 18877–18883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergqvist, A.; Rice, C.M. Transcriptional Activation of the Interleukin-2 Promoter by Hepatitis C Virus Core Protein. J. Virol. 2001, 75, 772–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moradpour, D.; Wakita, T.; Tokushige, K.; Carlson, R.I.; Krawczynski, K.; Wands, J.R. Characterization of three novel monoclonal antibodies against hepatitis C virus core protein. J. Med. Virol. 1996, 48, 234–241. [Google Scholar] [CrossRef]
- Hewitt, L.; Tighe, A.; Santaguida, S.; White, A.M.; Jones, C.D.; Musacchio, A.; Green, S.; Taylor, S.S. Sustained Mps1 activity is required in mitosis to recruit O-Mad2 to the Mad1–C-Mad2 core complex. J. Cell Biol. 2010, 190, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Northrop, J.P.; Ullman, K.S.; Crabtree, G.R. Characterization of the nuclear and cytoplasmic components of the lymphoid-specific nuclear factor of activated T cells (NF-AT) complex. J. Biol. Chem. 1993, 268, 2917–2923. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hwang, S.B.; Jeng, K.-S.; Zhu, N.; Lai, M.M. Homotypic Interaction and Multimerization of Hepatitis C Virus Core Protein. Virology 1996, 218, 43–51. [Google Scholar] [CrossRef] [Green Version]
- Majeau, N.; Gagné, V.; Boivin, A.; Bolduc, M.; Majeau, J.-A.; Ouellet, D.; Leclerc, D. The N-terminal half of the core protein of hepatitis C virus is sufficient for nucleocapsid formation. J. Gen. Virol. 2004, 85, 971–981. [Google Scholar] [CrossRef]
- Lorenzo, L.J.; Dueñas-Carrera, S.; Falcon, V.; Acosta-Rivero, N.; González, E.; de la Rosa, M.C.; Menéndez, I.; Morales, J. Assembly of Truncated HCV Core Antigen into Virus-like Particles in Escherichia coli. Biochem. Biophys. Res. Commun. 2001, 281, 962–965. [Google Scholar] [CrossRef]
- Kunkel, M.; Lorinczi, M.; Rijnbrand, R.; Lemon, S.M.; Watowich, S.J. Self-Assembly of Nucleocapsid-Like Particles from Recombinant Hepatitis C Virus Core Protein. J. Virol. 2001, 75, 2119–2129. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.; Ha, Y.; Park, H.-J. Structural requirements for assembly and homotypic interactions of the hepatitis C virus core protein. Virus Res. 2006, 122, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.C.; Dellos, S.R.; Lingappa, J.R. Identification of residues in the hepatitis C virus core protein that are critical for capsid assembly in a cell-free system. J. Virol. 2005, 79, 6814–6826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hope, R.G.; Mclauchlan, J. Sequence motifs required for lipid droplet association and protein stability are unique to the hepatitis C virus core protein. J. Gen. Virol. 2000, 81, 1913–1925. [Google Scholar] [CrossRef] [PubMed]
- Hourioux, C.; Ait-Goughoulte, M.; Patient, R.; Fouquenet, D.; Arcanger-Doudet, F.; Brand, D.; Martin, A.; Roingeard, P. Core protein domains involved in hepatitis C virus-like particle assembly and budding at the endoplasmic reticulum membrane. Cell. Microbiol. 2007, 9, 1014–1027. [Google Scholar] [CrossRef] [Green Version]
- Shavinskaya, A.; Boulant, S.; Penin, F.; McLauchlan, J.; Bartenschlager, R. The Lipid Droplet Binding Domain of Hepatitis C Virus Core Protein Is a Major Determinant for Efficient Virus Assembly. J. Biol. Chem. 2007, 282, 37158–37169. [Google Scholar] [CrossRef] [Green Version]
- Boulant, S.; Montserret, R.; Hope, R.G.; Ratinier, M.; Targett-Adams, P.; Lavergne, J.-P.; Penin, F.; McLauchlan, J. Structural Determinants That Target the Hepatitis C Virus Core Protein to Lipid Droplets. J. Biol. Chem. 2006, 281, 22236–22247. [Google Scholar] [CrossRef] [Green Version]
- Chang, S.C.; Yen, J.H.; Kang, H.Y.; Jang, M.H.; Chang, M.F. Nuclear Localization Signals in the Core Protein of Hepatitis C Virus. Biochem. Biophys. Res. Commun. 1994, 205, 1284–1290. [Google Scholar] [CrossRef]
- Suzuki, R.; Sakamoto, S.; Tsutsumi, T.; Rikimaru, A.; Tanaka, K.; Shimoike, T.; Moriishi, K.; Iwasaki, T.; Mizumoto, K.; Matsuura, Y.; et al. Molecular Determinants for Subcellular Localization of Hepatitis C Virus Core Protein. J. Virol. 2005, 79, 1271–1281. [Google Scholar] [CrossRef] [Green Version]
- Levin, A.; Neufeldt, C.J.; Pang, D.; Wilson, K.; Loewen-Dobler, D.; Joyce, M.A.; Wozniak, R.W.; Tyrrell, D.L.J. Functional Characterization of Nuclear Localization and Export Signals in Hepatitis C Virus Proteins and Their Role in the Membranous Web. PLoS ONE 2014, 9, e114629. [Google Scholar] [CrossRef]
- Blanchard, E.; Hourioux, C.; Brand, D.; Ait-Goughoulte, M.; Moreau, A.; Trassard, S.; Sizaret, P.-Y.; Dubois, F.; Roingeard, P. Hepatitis C Virus-Like Particle Budding: Role of the Core Protein and Importance of Its Asp111. J. Virol. 2003, 77, 10131–10138. [Google Scholar] [CrossRef] [Green Version]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef]
- Champeimont, R.; Laine, E.; Hu, S.-W.; Penin, F.; Carbone, A. Coevolution analysis of Hepatitis C virus genome to identify the structural and functional dependency network of viral proteins. Sci. Rep. 2016, 6, 26401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luik, P.; Chew, C.; Aittoniemi, J.; Chang, J.; Wentworth, P., Jr.; Dwek, R.A.; Biggin, P.C.; Vénien-Bryan, C.; Zitzmann, N. The 3-dimensional structure of a hepatitis C virus p7 ion channel by electron microscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 12712–12716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patargias, G.; Zitzmann, N.; Dwek, R.; Fischer, W.B. Protein−Protein Interactions: Modeling the Hepatitis C Virus Ion Channel p7. J. Med. Chem. 2006, 49, 648–655. [Google Scholar] [CrossRef] [Green Version]
- Chandler, D.E.; Penin, F.; Schulten, K.; Chipot, C. The p7 Protein of Hepatitis C Virus Forms Structurally Plastic, Minimalist Ion Channels. PLOS Comput. Biol. 2012, 8, e1002702. [Google Scholar] [CrossRef]
- Steinmann, E.; Pietschmann, T. Hepatitis C Virus P7—A Viroporin Crucial for Virus Assembly and an Emerging Target for Antiviral Therapy. Viruses 2010, 2, 2078–2095. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S.; Sato, N.; Kikuchi, J.; Kakinuma, H.; Okawa, J.; Masuyama, Y.; Iwasa, S.; Irokawa, H.; Hwang, G.-W.; Naganuma, A.; et al. Immature Core protein of hepatitis C virus induces an unfolded protein response through inhibition of ERAD-L in a yeast model system. Genes Cells 2017, 22, 160–173. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Plasmid | Sequence of Inserted Oligonucleotide | Enzyme Sites Used for Insertion |
|---|---|---|
| pHCVC∆2–12 | gaaGAATTCcaccATGcgtaacaccaaccgtcg | Eco RI a, Avr II a |
| pHCVC∆2–25 | ctaaGAATTCcaccATGggcggtcagatcgttg | Eco RI a, Avr II a |
| pHCVC∆26–57 | caaaCTCGAGgacccgggaacttgacg | Nde I b, Xho I |
| pHCVC∆61–68 | caaagCTCGAGgtcgtcggcccgagggcag | Eco RI a, Avr II a |
| pHCVC∆70–79 | caaGGTACCCacgtgccttggggatagg | Nde I b, Kpn I |
| pHCVC∆81–123 | cagccgg/cgatacc | Kpn I, Cla I |
| pHCVC∆124–142 | aggtcatcgaGCGGCCGCttggaggcgct | Cla I, Xag I |
| pHCVC∆147–161 | cgtcggcgccCCTcttggAGGcgtgaactatgcaacagg | Nde I b, Xag I |
| pHCVC1–161 | ctaaGAATTCagccgtcttccagaaccc | Eco RI a, Eco RI a |
| pHCVC1–173 | gaaGAATTCaagagcaaccaggaagg | Eco RI a, Eco RI a |
| pHCVC1–182 | gaaGAATTCagagcagggccagaagg | Eco RI a, Eco RI a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Devi, P.; Punga, T.; Bergqvist, A. Activation of the Ca2+/NFAT Pathway by Assembly of Hepatitis C Virus Core Protein into Nucleocapsid-like Particles. Viruses 2022, 14, 761. https://doi.org/10.3390/v14040761
Devi P, Punga T, Bergqvist A. Activation of the Ca2+/NFAT Pathway by Assembly of Hepatitis C Virus Core Protein into Nucleocapsid-like Particles. Viruses. 2022; 14(4):761. https://doi.org/10.3390/v14040761
Chicago/Turabian StyleDevi, Priya, Tanel Punga, and Anders Bergqvist. 2022. "Activation of the Ca2+/NFAT Pathway by Assembly of Hepatitis C Virus Core Protein into Nucleocapsid-like Particles" Viruses 14, no. 4: 761. https://doi.org/10.3390/v14040761
APA StyleDevi, P., Punga, T., & Bergqvist, A. (2022). Activation of the Ca2+/NFAT Pathway by Assembly of Hepatitis C Virus Core Protein into Nucleocapsid-like Particles. Viruses, 14(4), 761. https://doi.org/10.3390/v14040761