
Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein–Barr Virus-Associated Malignancies



Abstract
:1. Introduction
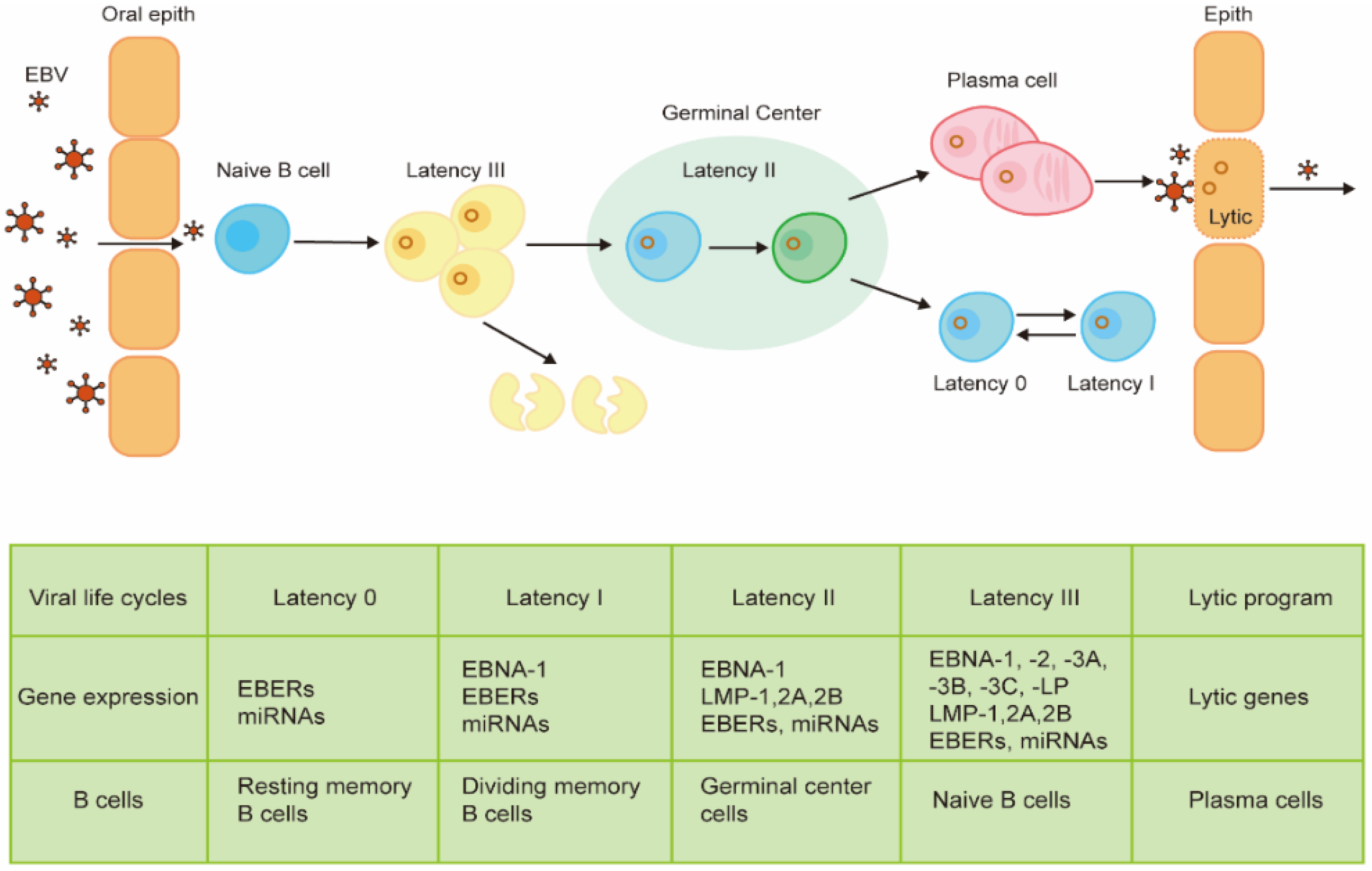
2. Viral Life Cycle and Gene Expression
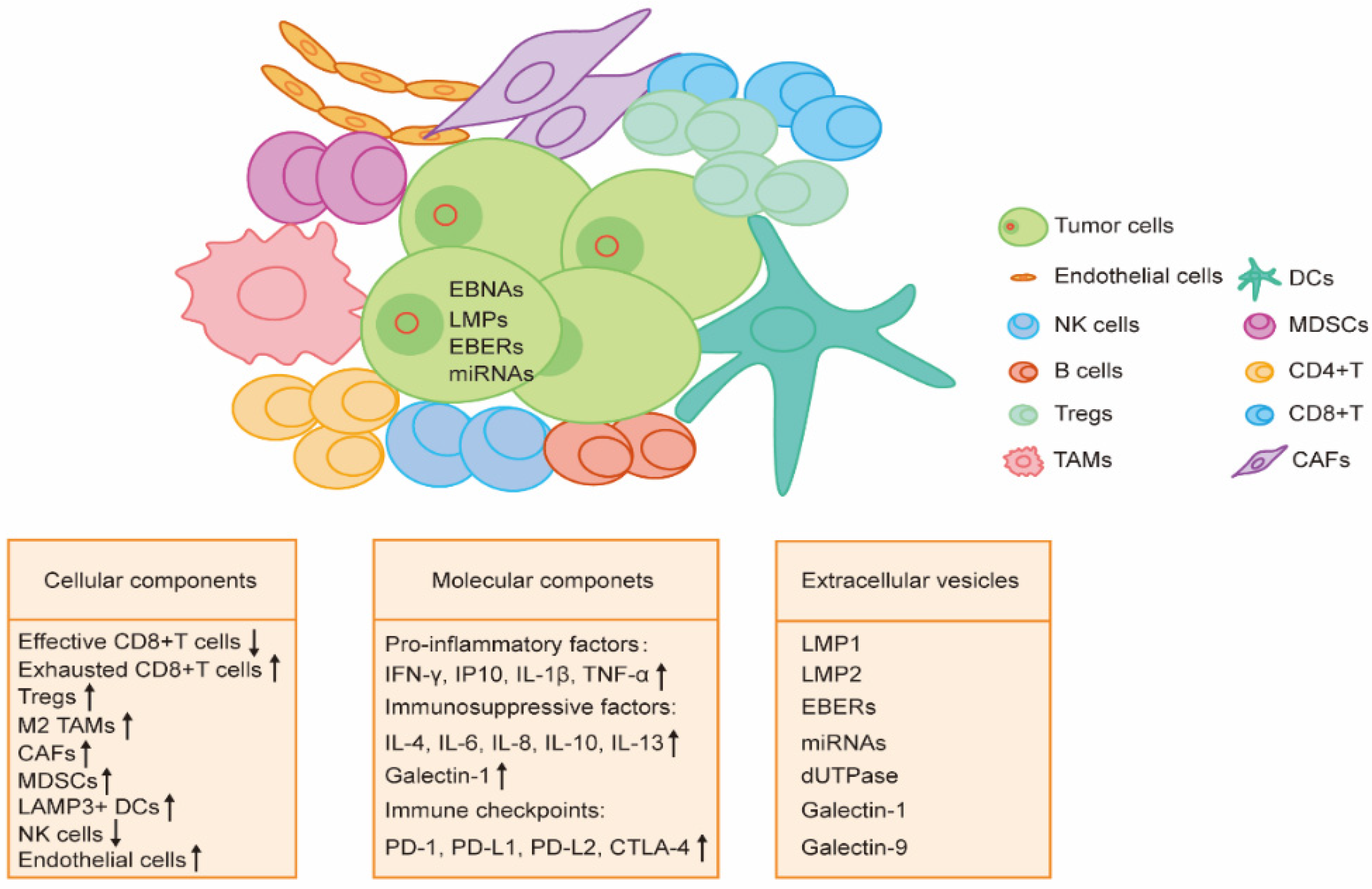
3. TME of EBV-Associated Malignancy
3.1. Cellular Components
3.1.1. T Cells
3.1.2. Tumor-Associated Macrophages (TAMs)
3.1.3. Dendritic Cells (DCs)
3.1.4. Myeloid-Derived Suppressor Cells (MDSCs)
3.1.5. NK Cells
3.1.6. B Cells
3.1.7. Cancer-Associated Fibroblasts (CAFs)
3.1.8. Endothelial Cells
3.2. Molecular Components
3.2.1. Soluble Molecules
3.2.2. Checkpoint Molecules
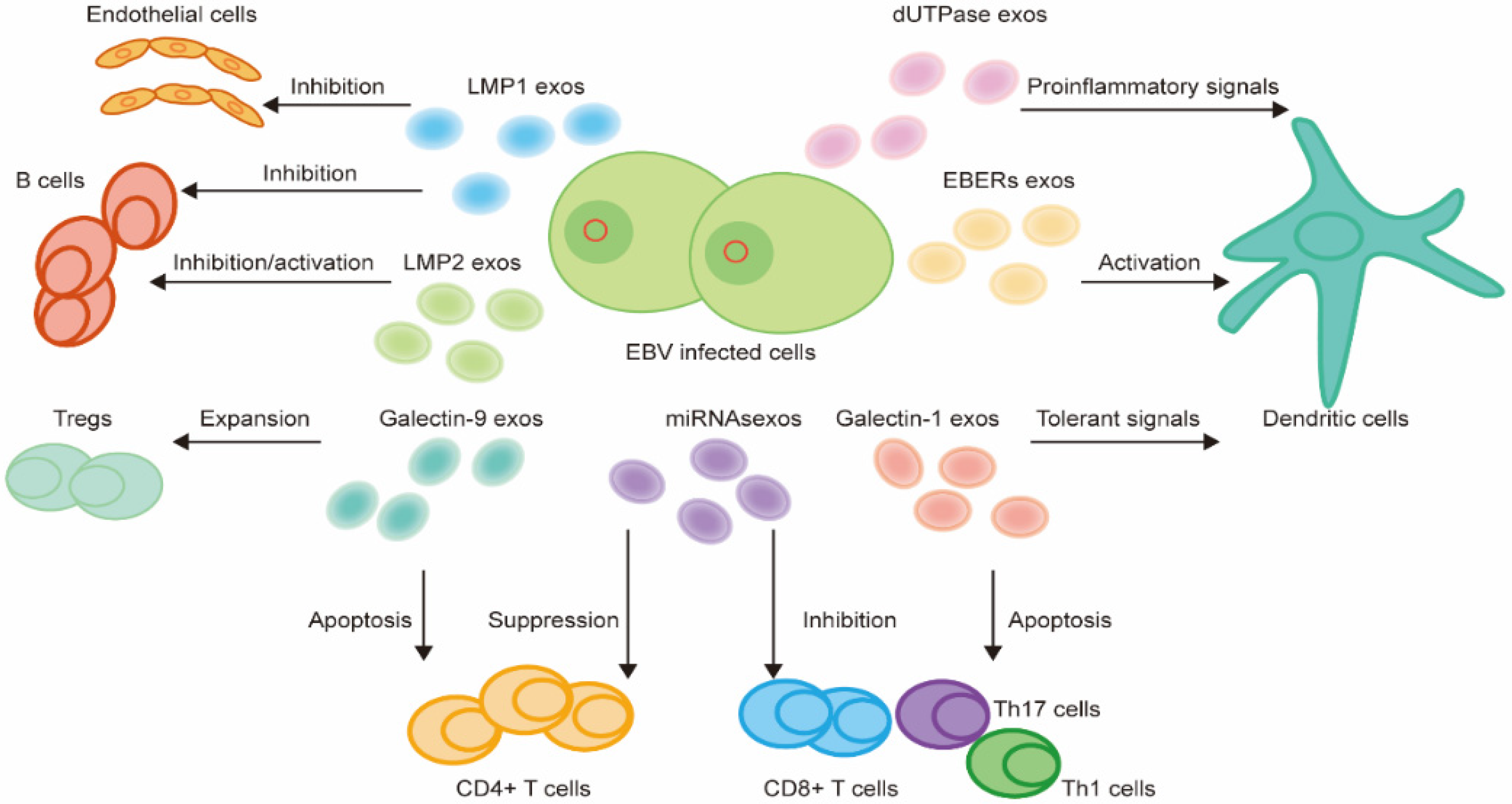
3.3. EVs
4. Landscape of Different EBV-Associated Malignancies
4.1. EBV-Associated Lymphomas
4.1.1. Post-transplant Lymphoproliferative Disorder (PTLD)
4.1.2. Hodgkin Lymphoma
4.1.3. Burkitt Lymphoma (BL)
4.1.4. Diffuse Large B Cell Lymphoma (DLBCL)
4.1.5. Extranodal NK/T Cell Lymphomas
4.2. NPC
4.3. EBVaGC
4.4. EBV-Associated Intrahepatic Cholangiocarcinoma (EBVaICC)
4.5. EBV-Associated Smooth Muscle Tumor (EBV-SMT)
5. Immunotherapy by Targeting the TME
5.1. Immune Checkpoint Inhibitors
5.2. T Cell Therapy
5.3. Therapeutic EBV Vaccine
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Epstein, A. Burkitt lymphoma and the discovery of Epstein-Barr virus. Br. J. Haematol. 2012, 156, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus particles in cultured lymphoblasts from Burkitt’s lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Wen, K.W.; Wang, L.; Menke, J.R.; Damania, B. Cancers associated with human gammaherpesviruses. FEBS J. 2021; pre-print. [Google Scholar] [CrossRef]
- Crawford, D.H.; Rickinson, A.; Johannessen, I.J.O.U.P. Cancer Virus: The story of Epstein-Barr Virus. Am. J. Epidemiol. 2014, 180, 1213–1215. [Google Scholar]
- Ressing, M.E.; van Gent, M.; Gram, A.M.; Hooykaas, M.J.; Piersma, S.J.; Wiertz, E.J. Immune Evasion by Epstein-Barr Virus. Curr. Top. Microbiol. Immunol. 2015, 391, 355–381. [Google Scholar]
- Machón, C.; Fàbrega-Ferrer, M.; Zhou, D.; Cuervo, A.; Carrascosa, J.L.; Stuart, D.I.; Coll, M. Atomic structure of the Epstein-Barr virus portal. Nat. Commun. 2019, 10, 3891. [Google Scholar] [CrossRef] [Green Version]
- Sample, J.; Young, L.; Martin, B.; Chatman, T.; Kieff, E.; Rickinson, A.; Kieff, E. Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J. Virol. 1990, 64, 4084–4092. [Google Scholar] [CrossRef] [Green Version]
- Dambaugh, T.; Hennessy, K.; Chamnankit, L.; Kieff, E. U2 region of Epstein-Barr virus DNA may encode Epstein-Barr nuclear antigen 2. Proc. Natl. Acad. Sci. USA 1984, 81, 7632–7636. [Google Scholar] [CrossRef] [Green Version]
- Zimber, U.; Adldinger, H.K.; Lenoir, G.M.; Vuillaume, M.; Knebel-Doeberitz, M.V.; Laux, G.; Desgranges, C.; Wittmann, P.; Freese, U.K.; Schneider, U. Geographical prevalence of two types of Epstein-Barr virus. Virology 1986, 154, 56–66. [Google Scholar] [CrossRef]
- Luzuriaga, K.; Sullivan, J.L. Infectious mononucleosis. N. Engl. J. Med. 2010, 362, 1993–2000. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef]
- Cesarman, E. Gammaherpesvirus and lymphoproliferative disorders in immunocompromised patients. Cancer Lett. 2011, 305, 163–174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesarman, E. Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. 2014, 9, 349–372. [Google Scholar] [CrossRef] [PubMed]
- de Martel, C.; Ferlay, J.; Franceschi, S.; Vignat, J.; Bray, F.; Forman, D.; Plummer, M. Global burden of cancers attributable to infections in 2008: A review and synthetic analysis. Lancet Oncol. 2012, 13, 607–615. [Google Scholar] [CrossRef]
- Khan, G.; Hashim, M.J. Global burden of deaths from Epstein-Barr virus attributable malignancies 1990–2010. Infect. Agents Cancer 2014, 9, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, G.; Fitzmaurice, C.; Naghavi, M.; Ahmed, L.A. Global and regional incidence, mortality and disability-adjusted life-years for Epstein-Barr virus-at tributable malignancies, 1990–2017. BMJ Open 2020, 10, e037505. [Google Scholar] [CrossRef]
- Tan, G.W.; Visser, L.; Tan, L.P.; van den Berg, A.; Diepstra, A. The Microenvironment in Epstein-Barr Virus-Associated Malignancies. Pathogens 2018, 7, 40. [Google Scholar] [CrossRef] [Green Version]
- Dolcetti, R. Cross-talk between Epstein-Barr virus and microenvironment in the pathogenesis of lymphomas. Semin. Cancer Biol. 2015, 34, 58–69. [Google Scholar] [CrossRef]
- Shen, Y.; Zhang, S.; Sun, R.; Wu, T.; Qian, J. Understanding the interplay between host immunity and Epstein-Barr virus in NPC patients. Emerg. Microbes Infect. 2015, 4, e20. [Google Scholar] [CrossRef]
- Iizasa, H.; Kim, H.; Kartika, A.V.; Kanehiro, Y.; Yoshiyama, H. Role of Viral and Host microRNAs in Immune Regulation of Epstein-Barr Virus-Associated Diseases. Front. Immunol. 2020, 11, 367. [Google Scholar] [CrossRef] [Green Version]
- Kintner, C.; Sugden, B. Conservation and progressive methylation of Epstein-Barr viral DNA sequences in transformed cells. J. Virol. 1981, 38, 305–316. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A.; Gross, A. Persistence of the Epstein-Barr virus and the origins of associated lymphomas. N. Engl. J. Med. 2004, 350, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Hochberg, D.; Thorley-Lawson, A.D. The expression pattern of Epstein-Barr virus latent genes in vivo is dependent upon the differentiati on stage of the infected B cell. Immunity 2000, 13, 497–506. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Woellmer, A.; Arteaga-Salas, J.M.; Hammerschmidt, W. BZLF1 governs CpG-methylated chromatin of Epstein-Barr Virus reversing epigenetic repression. PLoS Pathog. 2012, 8, e1002902. [Google Scholar] [CrossRef] [PubMed]
- Roughan, J.E.; Thorley-Lawson, D.A. The intersection of Epstein-Barr virus with the germinal center. J. Virol. 2009, 83, 3968–3976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caldwell, R.G.; Wilson, J.B.; Anderson, S.J.; Longnecker, R. Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell recep tor signals. Immunity 1998, 9, 405–411. [Google Scholar] [CrossRef] [Green Version]
- Gires, O.; Zimber-Strobl, U.; Gonnella, R.; Ueffing, M.; Marschall, G.; Zeidler, R.; Pich, D.; Hammerschmidt, W. Latent membrane protein 1 of Epstein-Barr virus mimics a constitutively active receptor molecule. EMBO J. 1997, 16, 6131–6140. [Google Scholar] [CrossRef] [Green Version]
- De Leo, A.; Calderon, A.; Lieberman, P.M. Control of Viral Latency by Episome Maintenance Proteins. Trends Microbiol. 2020, 28, 150–162. [Google Scholar] [CrossRef]
- Chiu, Y.F.; Sugden, B. Plasmid Partitioning by Human Tumor Viruses. J. Virol. 2018, 92, e02170-17. [Google Scholar] [CrossRef] [Green Version]
- Laichalk, L.L.; Thorley-Lawson, D.A. Terminal differentiation into plasma cells initiates the replicative cycle of Epstein-Barr virus in vivo. J. Virol. 2005, 79, 1296–1307. [Google Scholar] [CrossRef] [Green Version]
- Amon, W.; Binné, U.K.; Bryant, H.; Jenkins, P.J.; Karstegl, C.E.; Farrell, P.J. Lytic cycle gene regulation of Epstein-Barr virus. J. Virol. 2004, 78, 13460–13469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Countryman, J.; Miller, G. Activation of expression of latent Epstein-Barr herpesvirus after gene transfer with a small cloned s ubfragment of heterogeneous viral DNA. Proc. Natl. Acad. Sci. USA 1985, 82, 4085–4089. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, T.Y.; Chu, Y.Y.; Yang, Y.C.; Hsu, S.W.; Liu, S.T.; Chang, L.K. MCAF1 and Rta-activated BZLF1 transcription in Epstein-Barr virus. PLoS ONE 2014, 9, e90698. [Google Scholar] [CrossRef] [PubMed]
- Buschle, A.; Hammerschmidt, W. Epigenetic lifestyle of Epstein-Barr virus. Semin. Immunopathol. 2020, 42, 131–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef]
- Dolcetti, R.; Menezes, J. Epstein-Barr virus and undifferentiated nasopharyngeal carcinoma: New immunobiological and molecular insights on a long-standing etiopathogenic association. Adv. Cancer Res. 2003, 87, 127–157. [Google Scholar]
- Rickinson, A.B. Co-infections, inflammation and oncogenesis: Future directions for EBV research. Semin. Cancer Biol. 2014, 26, 99–115. [Google Scholar] [CrossRef]
- Cai, M.B.; Han, H.Q.; Bei, J.X.; Liu, C.C.; Lei, J.J.; Cui, Q.; Feng, Q.S.; Wang, H.Y.; Zhang, J.X.; Liang, Y.; et al. Expression of human leukocyte antigen G is associated with prognosis in nasopharyngeal carcinoma. Int. J. Biol. Sci. 2012, 8, 891–900. [Google Scholar] [CrossRef] [Green Version]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.; Keating, S.; Gomez, R.; Franken, K.L.; Ottenhoff, T.H.; Spriggs, M.; Schumacher, T.N.; Hutt-Fletcher, L.M.; et al. Epstein-Barr virus gp42 is posttranslationally modified to produce soluble gp42 that mediates HLA cla ss II immune evasion. J. Virol. 2005, 79, 841–852. [Google Scholar] [CrossRef] [Green Version]
- Ressing, M.E.; van Leeuwen, D.; Verreck, F.A.; Gomez, R.; Heemskerk, B.; Toebes, M.; Mullen, M.M.; Jardetzky, T.S.; Longnecker, R.; Schilham, M.W.; et al. Interference with T cell receptor-HLA-DR interactions by Epstein-Barr virus gp42 results in reduced T helper cell recognition. Proc. Natl. Acad. Sci. USA 2003, 100, 11583–11588. [Google Scholar] [CrossRef] [Green Version]
- Rowe, M.; Glaunsinger, B.; van Leeuwen, D.; Zuo, J.; Sweetman, D.; Ganem, D.; Middeldorp, J.; Wiertz, E.J.; Ressing, M.E. Host shutoff during productive Epstein-Barr virus infection is mediated by BGLF5 and may contribute to immune evasion. Proc. Natl. Acad. Sci. USA 2007, 104, 3366–3371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, D.G.; Trifilo, M.J.; Edelmann, K.H.; Teyton, L.; McGavern, D.B.; Oldstone, M.B. Interleukin-10 determines viral clearance or persistence in vivo. Nat. Med. 2006, 12, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Marshall, N.A.; Christie, L.E.; Munro, L.R.; Culligan, D.J.; Johnston, P.W.; Barker, R.N.; Vickers, M.A. Immunosuppressive regulatory T cells are abundant in the reactive lymphocytes of Hodgkin lymphoma. Blood 2004, 103, 1755–1762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baráth, S.; Aleksza, M.; Keresztes, K.; Tóth, J.; Sipka, S.; Szegedi, G.; Illés, A. Immunoregulatory T cells in the peripheral blood of patients with Hodgkin’s lymphoma. Acta Haematol. 2006, 116, 181–185. [Google Scholar] [CrossRef]
- Baumforth, K.R.; Birgersdotter, A.; Reynolds, G.M.; Wei, W.; Kapatai, G.; Flavell, J.R.; Kalk, E.; Piper, K.; Lee, S.; Machado, L.; et al. Expression of the Epstein-Barr virus-encoded Epstein-Barr virus nuclear antigen 1 in Hodgkin’s lympho ma cells mediates Up-regulation of CCL20 and the migration of regulatory T cells. Am. J. Pathol. 2008, 173, 195–204. [Google Scholar] [CrossRef] [Green Version]
- Marshall, N.A.; Culligan, D.J.; Tighe, J.; Johnston, P.W.; Barker, R.N.; Vickers, M.A. The relationships between Epstein-Barr virus latent membrane protein 1 and regulatory T cells in Hodg kin’s lymphoma. Exp. Hematol. 2007, 35, 596–604. [Google Scholar] [CrossRef]
- Morales, O.; Mrizak, D.; François, V.; Mustapha, R.; Miroux, C.; Depil, S.; Decouvelaere, A.V.; Lionne-Huyghe, P.; Auriault, C.; de Launoit, Y.; et al. Epstein-Barr virus infection induces an increase of T regulatory type 1 cells in Hodgkin lymphoma pat ients. Br. J. Haematol. 2014, 166, 875–890. [Google Scholar] [CrossRef]
- Barros, M.H.; Segges, P.; Vera-Lozada, G.; Hassan, R.; Niedobitek, G. Macrophage polarization reflects T cell composition of tumor microenvironment in pediatric classical Hodgkin lymphoma and has impact on survival. PLoS ONE 2015, 10, e0124531. [Google Scholar] [CrossRef]
- Buettner, M.; Meyer, B.; Schreck, S.; Niedobitek, G. Expression of RANTES and MCP-1 in epithelial cells is regulated via LMP1 and CD40. Int. J. Cancer 2007, 121, 2703–2710. [Google Scholar] [CrossRef]
- Huang, H.; Liu, X.; Zhao, F.; Lu, J.; Zhang, B.; Peng, X.; Zhang, M.; Chen, X.; Li, G.; Li, X. M2-polarized tumour-associated macrophages in stroma correlate with poor prognosis and Epstein-Barr v iral infection in nasopharyngeal carcinoma. Acta Otolaryngol. 2017, 137, 888–894. [Google Scholar] [CrossRef]
- Steinman, R.M. The dendritic cell system and its role in immunogenicity. Annu. Rev. Immunol. 1991, 9, 271–296. [Google Scholar] [CrossRef] [PubMed]
- Banchereau, J.; Steinman, R.M. Dendritic cells and the control of immunity. Nature 1998, 392, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Chen, X.M.; Huang, H.R.; Zhao, F.P.; Wang, F.; Liu, X.; Li, X.P. Detailed analysis of inflammatory cell infiltration and the prognostic impact on nasopharyngeal carci noma. Head Neck 2018, 40, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Braz-Silva, P.H.; Vitale, S.; Butori, C.; Guevara, N.; Santini, J.; Magalhães, M.; Hofman, P.; Doglio, A. Specific infiltration of langerin-positive dendritic cells in EBV-infected tonsil, Hodgkin lymphoma and nasopharyngeal carcinoma. Int. J. Cancer 2011, 128, 2501–2508. [Google Scholar] [CrossRef]
- Ariza, M.E.; Rivailler, P.; Glaser, R.; Chen, M.; Williams, M.V. Epstein-Barr virus encoded dUTPase containing exosomes modulate innate and adaptive immune responses in human dendritic cells and peripheral blood mononuclear cells. PLoS ONE 2013, 8, e69827. [Google Scholar] [CrossRef] [Green Version]
- Hinata, M.; Kunita, A.; Abe, H.; Morishita, Y.; Sakuma, K.; Yamashita, H.; Seto, Y.; Ushiku, T.; Fukayama, M. Exosomes of Epstein-Barr Virus-Associated Gastric Carcinoma Suppress Dendritic Cell Maturation. Microorganisms 2020, 8, 1776. [Google Scholar] [CrossRef]
- Liu, Y.; He, S.; Wang, X.L.; Peng, W.; Chen, Q.Y.; Chi, D.M.; Chen, J.R.; Han, B.W.; Lin, G.W.; Li, Y.Q.; et al. Tumour heterogeneity and intercellular networks of nasopharyngeal carcinoma at single cell resolution. Nat. Commun. 2021, 12, 741. [Google Scholar] [CrossRef]
- Oh, S.A.; Wu, D.C.; Cheung, J.; Navarro, A.; Xiong, H.; Cubas, R.; Totpal, K.; Chiu, H.; Wu, Y.; Comps-Agrar, L.; et al. PD-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat. Cancer 2020, 1, 681–691. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Cai, T.T.; Ye, S.B.; Liu, Y.N.; He, J.; Chen, Q.Y.; Mai, H.Q.; Zhang, C.X.; Cui, J.; Zhang, X.S.; Busson, P.; et al. LMP1-mediated glycolysis induces myeloid-derived suppressor cell expansion in nasopharyngeal carcinoma. PLoS Pathog. 2017, 13, e1006503. [Google Scholar] [CrossRef]
- Hoechst, B.; Gamrekelashvili, J.; Manns, M.P.; Greten, T.F.; Korangy, F. Plasticity of human Th17 cells and iTregs is orchestrated by different subsets of myeloid cells. Blood 2011, 117, 6532–6541. [Google Scholar] [CrossRef] [PubMed]
- Vetsika, E.K.; Koukos, A.; Kotsakis, A. Myeloid-Derived Suppressor Cells: Major Figures that Shape the Immunosuppressive and Angiogenic Network in Cancer. Cells 2019, 8, 1647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlecker, E.; Stojanovic, A.; Eisen, C.; Quack, C.; Falk, C.S.; Umansky, V.; Cerwenka, A. Tumor-infiltrating monocytic myeloid-derived suppressor cells mediate CCR5-dependent recruitment of regulatory T cells favoring tumor growth. J. Immunol. 2012, 189, 5602–5611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abbott, R.J.; Pachnio, A.; Pedroza-Pacheco, I.; Leese, A.M.; Begum, J.; Long, H.M.; Croom-Carter, D.; Stacey, A.; Moss, P.A.H.; Hislop, A.D.; et al. Asymptomatic Primary Infection with Epstein-Barr Virus: Observations on Young Adult Cases. J. Virol. 2017, 91, e00382-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, H.; McAulay, K.; Macsween, K.F.; Gallacher, N.J.; Higgins, C.D.; Harrison, N.; Swerdlow, A.J.; Crawford, D.H. The immune response to primary EBV infection: A role for natural killer cells. Br. J. Haematol. 2005, 129, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Lo, K.W.; To, K.F.; Huang, D.P. Focus on nasopharyngeal carcinoma. Cancer Cell 2004, 5, 423–428. [Google Scholar] [CrossRef] [Green Version]
- Everett, H.; McFadden, G. Viruses and apoptosis: Meddling with mitochondria. Virology 2001, 288, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Yoshimori, M.; Imadome, K.; Komatsu, H.; Wang, L.; Saitoh, Y.; Yamaoka, S.; Fukuda, T.; Kurata, M.; Koyama, T.; Shimizu, N.; et al. CD137 expression is induced by Epstein-Barr virus infection through LMP1 in T or NK cells and mediate s survival promoting signals. PLoS ONE 2014, 9, e112564. [Google Scholar] [CrossRef]
- Vidard, L.; Dureuil, C.; Baudhuin, J.; Vescovi, L.; Durand, L.; Sierra, V.; Parmantier, E. CD137 (4-1BB) Engagement Fine-Tunes Synergistic IL-15- and IL-21-Driven NK Cell Proliferation. J. Immunol. 2019, 203, 676–685. [Google Scholar] [CrossRef]
- Makowska, A.; Braunschweig, T.; Denecke, B.; Shen, L.; Baloche, V.; Busson, P.; Kontny, U. Interferon β and Anti-PD-1/PD-L1 Checkpoint Blockade Cooperate in NK Cell-Mediated Killing of Nasopharyngeal Carcinoma Cells. Transl. Oncol. 2019, 12, 1237–1256. [Google Scholar] [CrossRef]
- Shah, K.M.; Stewart, S.E.; Wei, W.; Woodman, C.B.; O’Neil, J.D.; Dawson, C.W.; Young, L.S. The EBV-encoded latent membrane proteins, LMP2A and LMP2B, limit the actions of interferon by targeting interferon receptors for degradation. Oncogene 2009, 28, 3903–3914. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, T.S.; Chen, S.; Zhang, M.J.; Chan, J.Y.; Gao, W. Epstein-Barr virus-encoded microRNA BART7 downregulates major histocompatibility complex class I chain-related peptide A and reduces the cytotoxicity of natural killer cells to nasopharyngeal carcinoma. Oncol. Lett. 2018, 16, 2887–2892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strowig, T.; Brilot, F.; Arrey, F.; Bougras, G.; Thomas, D.; Muller, W.A.; Münz, C. Tonsilar NK cells restrict B cell transformation by the Epstein-Barr virus via IFN-gamma. PLoS Pathog. 2008, 4, e27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Png, Y.T.; Yang, A.Z.Y.; Lee, M.Y.; Chua, M.J.M.; Lim, C.M. The Role of NK Cells in EBV Infection and EBV-Associated NPC. Viruses 2021, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Zong, Y.S.; Lin, H.; Choy, D.T.; Sham, J.S.; Wei, W.; Chan, K.H.; Ng, M.H. Nasopharyngeal carcinoma and lymphoinfiltration. Oncology 1991, 48, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Sakakibara, S.; Yasui, T.; Minamitani, T.; Okuzaki, D.; Kikutani, H. Bystander inhibition of humoral immune responses by Epstein-Barr virus LMP1. Int. Immunol. 2018, 30, 579–590. [Google Scholar] [CrossRef]
- Miao, B.P.; Zhang, R.S.; Li, M.; Fu, Y.T.; Zhao, M.; Liu, Z.G.; Yang, P.C. Nasopharyngeal cancer-derived microRNA-21 promotes immune suppressive B cells. Cell Mol. Immunol. 2015, 12, 750–756. [Google Scholar] [CrossRef]
- Wu, X.; Zhou, Z.; Xu, S.; Liao, C.; Chen, X.; Li, B.; Peng, J.; Li, D.; Yang, L. Extracellular vesicle packaged LMP1-activated fibroblasts promote tumor progression via autophagy and stroma-tumor metabolism coupling. Cancer Lett. 2020, 478, 93–106. [Google Scholar] [CrossRef]
- Davis, A.M.; Rapley, A.; Dawson, C.W.; Young, L.S.; Morris, M.A. The EBV-Encoded Oncoprotein, LMP1, Recruits and Transforms Fibroblasts via an ERK-MAPK-Dependent Mechanism. Pathogens 2021, 10, 982. [Google Scholar] [CrossRef]
- Yamamura, Y.; Asai, N.; Enomoto, A.; Kato, T.; Mii, S.; Kondo, Y.; Ushida, K.; Niimi, K.; Tsunoda, N.; Nagino, M.; et al. Akt-Girdin signaling in cancer-associated fibroblasts contributes to tumor progression. Cancer Res. 2015, 75, 813–823. [Google Scholar] [CrossRef] [Green Version]
- Orimo, A.; Gupta, P.B.; Sgroi, D.C.; Arenzana-Seisdedos, F.; Delaunay, T.; Naeem, R.; Carey, V.J.; Richardson, A.L.; Weinberg, R.A. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell 2005, 121, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Niu, Y.; Yeh, S.; Chang, C. BM-MSCs promote prostate cancer progression via the conversion of normal fibroblasts to cancer-associated fibroblasts. Int. J. Oncol. 2015, 47, 719–727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michiels, C. Endothelial cell functions. J. Cell. Physiol. 2003, 196, 430–443. [Google Scholar] [CrossRef] [PubMed]
- Murono, S.; Inoue, H.; Tanabe, T.; Joab, I.; Yoshizaki, T.; Furukawa, M.; Pagano, J.S. Induction of cyclooxygenase-2 by Epstein-Barr virus latent membrane protein 1 is involved in vascular endothelial growth factor production in nasopharyngeal carcinoma cells. Proc. Natl. Acad. Sci. USA 2001, 98, 6905–6910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Li, Z.; He, J.; Fu, S.; Duan, Y.; Zhou, Q.; Yan, Y.; Liu, X.; Liu, L.; Feng, C.; et al. Epstein-Barr virus noncoding RNAs from the extracellular vesicles of nasopharyngeal carcinoma (NPC) cells promote angiogenesis via TLR3/RIG-I-mediated VCAM-1 expression. Biochimica et biophysica acta. Mol. Basis Dis. 2019, 1865, 1201–1213. [Google Scholar] [CrossRef]
- Farina, A.; Rosato, E.; York, M.; Gewurz, B.E.; Trojanowska, M.; Farina, G.A. Innate Immune Modulation Induced by EBV Lytic Infection Promotes Endothelial Cell Inflammation and Vascular Injury in Scleroderma. Front. Immunol. 2021, 12, 651013. [Google Scholar] [CrossRef]
- Maggio, E.; van den Berg, A.; Diepstra, A.; Kluiver, J.; Visser, L.; Poppema, S. Chemokines, cytokines and their receptors in Hodgkin’s lymphoma cell lines and tissues. Ann. Oncol. 2002, 13 (Suppl. 1), 52–56. [Google Scholar] [CrossRef]
- Liu, M.; Guo, S.; Stiles, J.K. The emerging role of CXCL10 in cancer (Review). Oncol. Lett. 2011, 2, 583–589. [Google Scholar] [CrossRef] [Green Version]
- Vockerodt, M.; Pinkert, D.; Smola-Hess, S.; Michels, A.; Ransohoff, R.M.; Tesch, H.; Kube, D. The Epstein-Barr virus oncoprotein latent membrane protein 1 induces expression of the chemokine IP-10: Importance of mRNA half-life regulation. Int. J. Cancer 2005, 114, 598–605. [Google Scholar] [CrossRef]
- Gong, L.P.; Chen, J.N.; Xiao, L.; He, Q.; Feng, Z.Y.; Zhang, Z.G.; Liu, J.P.; Wei, H.B.; Shao, C.K. The implication of tumor-infiltrating lymphocytes in Epstein-Barr virus-associated gastric carcinoma. Hum. Pathol. 2019, 85, 82–91. [Google Scholar] [CrossRef]
- Chen, L.C.; Wang, L.J.; Tsang, N.M.; Ojcius, D.M.; Chen, C.C.; Ouyang, C.N.; Hsueh, C.; Liang, Y.; Chang, K.P.; Chen, C.C.; et al. Tumour inflammasome-derived IL-1β recruits neutrophils and improves local recurrence-free survival in EBV-induced nasopharyngeal carcinoma. EMBO Mol. Med. 2012, 4, 1276–1293. [Google Scholar] [CrossRef] [PubMed]
- Chuang, H.C.; Lay, J.D.; Chuang, S.E.; Hsieh, W.C.; Chang, Y.; Su, I.J. Epstein-Barr virus (EBV) latent membrane protein-1 down-regulates tumor necrosis factor-alpha (TNF-alpha) receptor-1 and confers resistance to TNF-alpha-induced apoptosis in T cells: Implication for the progression to T-cell lymphoma in EBV-associated hemophagocytic syndrome. Am. J. Pathol. 2007, 170, 1607–1617. [Google Scholar] [PubMed] [Green Version]
- Micheau, O.; Tschopp, J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell 2003, 114, 181–190. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Yang, J.; Li, M.; Huang, N.; Chen, Y.; Wu, X.; Wang, X.; Qiu, S.; Wang, H.; Li, X. Viral IL-10 promotes cell proliferation and cell cycle progression via JAK2/STAT3 signaling pathway in nasopharyngeal carcinoma cells. Biotechnol. Appl. Biochem. 2020, 67, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Roncarolo, M.G.; Bacchetta, R.; Bordignon, C.; Narula, S.; Levings, M.K. Type 1 T regulatory cells. Immunol. Rev. 2001, 182, 68–79. [Google Scholar] [CrossRef] [PubMed]
- Pachnia, D.; Drop, B.; Dworzańska, A.; Kliszczewska, E.; Polz-Dacewicz, M. Transforming Growth Factor-β, Interleukin-10, and Serological Markers in EBV-associated Gastric Carcinoma. Anticancer Res. 2017, 37, 4853–4858. [Google Scholar] [PubMed]
- Yao, M.; Ohshima, K.; Suzumiya, J.; Kume, T.; Shiroshita, T.; Kikuchi, M. Interleukin-10 expression and cytotoxic-T-cell response in Epstein-Barr-virus-associated nasopharyngeal carcinoma. Int. J. Cancer 1997, 72, 398–402. [Google Scholar] [CrossRef]
- Incrocci, R.; Barse, L.; Stone, A.; Vagvala, S.; Montesano, M.; Subramaniam, V.; Swanson-Mungerson, M. Epstein-Barr Virus Latent Membrane Protein 2A (LMP2A) enhances IL-10 production through the activation of Bruton’s tyrosine kinase and STAT3. Virology 2017, 500, 96–102. [Google Scholar] [CrossRef]
- Sakamoto, T.; Saito, H.; Tatebe, S.; Tsujitani, S.; Ozaki, M.; Ito, H.; Ikeguchi, M. Interleukin-10 expression significantly correlates with minor CD8+ T-cell infiltration and high microvessel density in patients with gastric cancer. Int. J. Cancer 2006, 118, 1909–1914. [Google Scholar] [CrossRef] [Green Version]
- Fior, R.; Vita, N.; Raphael, M.; Minty, A.; Maillot, M.C.; Crevon, M.C.; Caput, D.; Biberfeld, P.; Ferrara, P.; Galanaud, P. Interleukin-13 gene expression by malignant and EBV-transformed human B lymphocytes. Eur. Cytokine Netw. 1994, 5, 593–600. [Google Scholar]
- Kis, L.L.; Gerasimcik, N.; Salamon, D.; Persson, E.K.; Nagy, N.; Klein, G.; Severinson, E.; Klein, E. STAT6 signaling pathway activated by the cytokines IL-4 and IL-13 induces expression of the Epstein-B arr virus-encoded protein LMP-1 in absence of EBNA-2: Implications for the type II EBV latent gene expression in Hodgkin lymphoma. Blood 2011, 117, 165–174. [Google Scholar] [CrossRef]
- Eliopoulos, A.G.; Stack, M.; Dawson, C.W.; Kaye, K.M.; Hodgkin, L.; Sihota, S.; Rowe, M.; Young, L.S. Epstein-Barr virus-encoded LMP1 and CD40 mediate IL-6 production in epithelial cells via an NF-kappaB pathway involving TNF receptor-associated factors. Oncogene 1997, 14, 2899–2916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eliopoulos, A.G.; Gallagher, N.J.; Blake, S.M.; Dawson, C.W.; Young, L.S. Activation of the p38 mitogen-activated protein kinase pathway by Epstein-Barr virus-encoded latent membrane protein 1 coregulates interleukin-6 and interleukin-8 production. J. Biol. Chem. 1999, 274, 16085–16096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, J.; Juszczynski, P.; Rodig, S.J.; Green, M.R.; O’Donnell, E.; Currie, T.; Armant, M.; Takeyama, K.; Monti, S.; Rabinovich, G.A.; et al. Viral induction and targeted inhibition of galectin-1 in EBV+ posttransplant lymphoproliferative disorders. Blood 2011, 117, 4315–4322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Zhang, W. Expression of PD-L1 in EBV-associated malignancies. Int. Immunopharmacol. 2021, 95, 107553. [Google Scholar] [CrossRef]
- Tsirigotis, P.; Savani, B.N.; Nagler, A. Programmed death-1 immune checkpoint blockade in the treatment of hematological malignancies. Ann. Med. 2016, 48, 428–439. [Google Scholar] [CrossRef]
- Ansell, S.M.; Lesokhin, A.M.; Borrello, I.; Halwani, A.; Scott, E.C.; Gutierrez, M.; Schuster, S.J.; Millenson, M.M.; Cattry, D.; Freeman, G.J.; et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N. Engl. J. Med. 2015, 372, 311–319. [Google Scholar] [CrossRef] [Green Version]
- Green, M.R.; Rodig, S.; Juszczynski, P.; Ouyang, J.; Sinha, P.; O’Donnell, E.; Neuberg, D.; Shipp, M.A. Constitutive AP-1 activity and EBV infection induce PD-L1 in Hodgkin lymphomas and posttransplant lymphoproliferative disorders: Implications for targeted therapy. Clin. Cancer Res. 2012, 18, 1611–1618. [Google Scholar] [CrossRef] [Green Version]
- Gilardini Montani, M.S.; Santarelli, R.; Falcinelli, L.; Gonnella, R.; Granato, M.; Di Renzo, L.; Cuomo, L.; Vitillo, M.; Faggioni, A.; Cirone, M. EBV up-regulates PD-L1 on the surface of primary monocytes by increasing ROS and activating TLR signa ling and STAT3. J. Leukoc. Biol. 2018, 104, 821–832. [Google Scholar] [CrossRef]
- Sasaki, S.; Nishikawa, J.; Sakai, K.; Iizasa, H.; Yoshiyama, H.; Yanagihara, M.; Shuto, T.; Shimokuri, K.; Kanda, T.; Suehiro, Y.; et al. EBV-associated gastric cancer evades T-cell immunity by PD-1/PD-L1 interactions. Gastric Cancer 2019, 22, 486–496. [Google Scholar] [CrossRef] [Green Version]
- Greenough, T.C.; Campellone, S.C.; Brody, R.; Jain, S.; Sanchez-Merino, V.; Somasundaran, M.; Luzuriaga, K. Programmed Death-1 expression on Epstein Barr virus specific CD8+ T cells varies by stage of infection, epitope specificity, and T-cell receptor usage. PLoS ONE 2010, 5, e12926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.M.; Gebriel, M.G.; Morad, E.A.; Saber, I.M.; Elwan, A.; Salah, M.; Fakhr, A.E.; Shalaby, A.M.; Alabiad, M.A. Expression of Immune Checkpoint Regulators, Cytotoxic T-Lymphocyte Antigen-4, and Programmed Death-Ligand 1 in Epstein-Barr Virus-associated Nasopharyngeal Carcinoma. Appl. Immunohistochem. Mol. Morphol. AIMM 2021, 29, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Q.; Zhang, Y.; Jiang, W.; Chen, Y.P.; Xu, S.Y.; Liu, N.; Zhao, Y.; Li, L.; Lei, Y.; Hong, X.H.; et al. Development and validation of an immune checkpoint-based signature to predict prognosis in nasopharyngeal carcinoma using computational pathology analysis. J. Immunother. Cancer 2019, 7, 298. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, M.K.; Lambley, E.; Duraiswamy, J.; Dua, U.; Smith, C.; Elliott, S.; Gill, D.; Marlton, P.; Seymour, J.; Khanna, R. Expression of LAG-3 by tumor-infiltrating lymphocytes is coincident with the suppression of latent membrane antigen-specific CD8+ T-cell function in Hodgkin lymphoma patients. Blood 2006, 108, 2280–2289. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G. Exosomal communication goes viral. J. Virol. 2015, 89, 5200–5203. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Cone, A.S.; York, S.B.; Meckes, D.G. Extracellular Vesicles in Epstein-Barr Virus Pathogenesis. Curr. Clin. Microbiol. Rep. 2019, 6, 121–131. [Google Scholar] [CrossRef]
- Meckes, D.G.; Raab-Traub, N. Microvesicles and viral infection. J. Virol. 2011, 85, 12844–12854. [Google Scholar] [CrossRef] [Green Version]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef] [Green Version]
- Gutzeit, C.; Nagy, N.; Gentile, M.; Lyberg, K.; Gumz, J.; Vallhov, H.; Puga, I.; Klein, E.; Gabrielsson, S.; Cerutti, A.; et al. Exosomes derived from Burkitt’s lymphoma cell lines induce proliferation, differentiation, and class- switch recombination in B cells. J. Immunol. 2014, 192, 5852–5862. [Google Scholar] [CrossRef] [Green Version]
- Fang, W.; Zhang, J.; Hong, S.; Zhan, J.; Chen, N.; Qin, T.; Tang, Y.; Zhang, Y.; Kang, S.; Zhou, T.; et al. EBV-driven LMP1 and IFN-γ up-regulate PD-L1 in nasopharyngeal carcinoma: Implications for oncotargeted therapy. Oncotarget 2014, 5, 12189–12202. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, S.; Visco, V.; Raffa, S.; Wakisaka, N.; Pagano, J.S.; Torrisi, M.R. Epstein-Barr virus latent membrane protein 1 promotes concentration in multivesicular bodies of fibroblast growth factor 2 and its release through exosomes. Int. J. Cancer 2007, 121, 1494–1506. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G.; Shair, K.H.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Longnecker, R. Cholesterol is critical for Epstein-Barr virus latent membrane protein 2A trafficking and protein stability. Virology 2007, 360, 461–468. [Google Scholar] [CrossRef]
- Cen, O.; Longnecker, R. Latent Membrane Protein 2 (LMP2). Curr. Top. Microbiol. Immunol. 2015, 391, 151–180. [Google Scholar]
- Ahmed, W.; Tariq, S.; Khan, G. Tracking EBV-encoded RNAs (EBERs) from the nucleus to the excreted exosomes of B-lymphocytes. Sci. Rep. 2018, 8, 15438. [Google Scholar] [CrossRef]
- Iwakiri, D. Epstein-Barr Virus-Encoded RNAs: Key Molecules in Viral Pathogenesis. Cancers 2014, 6, 1615–1630. [Google Scholar] [CrossRef] [Green Version]
- Takada, K. Role of EBER and BARF1 in nasopharyngeal carcinoma (NPC) tumorigenesis. Semin. Cancer Biol. 2012, 22, 162–165. [Google Scholar] [CrossRef]
- Iwakiri, D.; Zhou, L.; Samanta, M.; Matsumoto, M.; Ebihara, T.; Seya, T.; Imai, S.; Fujieda, M.; Kawa, K.; Takada, K. Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3. J. Exp. Med. 2009, 206, 2091–2099. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Meckes, D.G. Methodological Approaches to Study Extracellular Vesicle miRNAs in Epstein–Barr Virus-Associated Cancers. Int. J. Mol. Sci. 2018, 19, 2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.J.; Huang, T.J.; Yang, C.F.; Peng, L.X.; Liu, R.Y.; Yang, G.D.; Chu, Q.Q.; Huang, J.L.; Liu, N.; Huang, H.B.; et al. Comprehensive profiling of Epstein-Barr virus-encoded miRNA species associated with specific latency types in tumor cells. Virol. J. 2013, 10, 314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tagawa, T.; Albanese, M.; Bouvet, M.; Moosmann, A.; Mautner, J.; Heissmeyer, V.; Zielinski, C.; Lutter, D.; Hoser, J.; Hastreiter, M.; et al. Epstein-Barr viral miRNAs inhibit antiviral CD4+ T cell responses targeting IL-12 and peptide processing. J. Exp. Med. 2016, 213, 2065–2080. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.A.; Bianco, G.A.; Ilarregui, J.M.; Croci, D.O.; Correale, J.; Hernandez, J.D.; Zwirner, N.W.; Poirier, F.; Riley, E.M.; Baum, L.G.; et al. Differential glycosylation of TH1, TH2 and TH-17 effector cells selectively regulates susceptibility to cell death. Nat. Immunol. 2007, 8, 825–834. [Google Scholar] [CrossRef]
- Perone, M.J.; Larregina, A.T.; Shufesky, W.J.; Papworth, G.D.; Sullivan, M.L.; Zahorchak, A.F.; Stolz, D.B.; Baum, L.G.; Watkins, S.C.; Thomson, A.W.; et al. Transgenic galectin-1 induces maturation of dendritic cells that elicit contrasting responses in naive and activated T cells. J. Immunol. 2006, 176, 7207–7220. [Google Scholar] [CrossRef] [Green Version]
- Keryer-Bibens, C.; Pioche-Durieu, C.; Villemant, C.; Souquère, S.; Nishi, N.; Hirashima, M.; Middeldorp, J.; Busson, P. Exosomes released by EBV-infected nasopharyngeal carcinoma cells convey the viral latent membrane protein 1 and the immunomodulatory protein galectin 9. BMC Cancer 2006, 6, 283. [Google Scholar] [CrossRef] [Green Version]
- Klibi, J.; Niki, T.; Riedel, A.; Pioche-Durieu, C.; Souquere, S.; Rubinstein, E.; Le Moulec, S.; Moulec, S.L.; Guigay, J.; Hirashima, M.; et al. Blood diffusion and Th1-suppressive effects of galectin-9-containing exosomes released by Epstein-Bar r virus-infected nasopharyngeal carcinoma cells. Blood 2009, 113, 1957–1966. [Google Scholar] [CrossRef] [Green Version]
- Mrizak, D.; Martin, N.; Barjon, C.; Jimenez-Pailhes, A.S.; Mustapha, R.; Niki, T.; Guigay, J.; Pancré, V.; de Launoit, Y.; Busson, P.; et al. Effect of nasopharyngeal carcinoma-derived exosomes on human regulatory T cells. J. Natl. Cancer Inst. 2015, 107, 363. [Google Scholar] [CrossRef] [Green Version]
- Ramayanti, O.; Verkuijlen, S.; Novianti, P.; Scheepbouwer, C.; Misovic, B.; Koppers-Lalic, D.; van Weering, J.; Beckers, L.; Adham, M.; Martorelli, D.; et al. Vesicle-bound EBV-BART13-3p miRNA in circulation distinguishes nasopharyngeal from other head and neck cancer and asymptomatic EBV-infections. Int. J. Cancer 2019, 144, 2555–2566. [Google Scholar] [CrossRef]
- Tsai, C.Y.; Liu, Y.Y.; Liu, K.H.; Hsu, J.T.; Chen, T.C.; Chiu, C.T.; Yeh, T.S. Comprehensive profiling of virus microRNAs of Epstein-Barr virus-associated gastric carcinoma: Highlighting the interactions of ebv-Bart9 and host tumor cells. J. Gastroenterol. Hepatol. 2017, 32, 82–91. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Xiang, Z.; Liu, Y.; Huang, C.; Pei, Y.; Wang, X.; Zhi, H.; Wong, W.H.; Wei, H.; Ng, I.O.; et al. Exosomes derived from Vδ2-T cells control Epstein-Barr virus-associated tumors and induce T cell antitumor immunity. Sci. Transl. Med. 2020, 12, eaaz3426. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Xie, Y.; Wang, T.; Wang, L. New insights into Epstein-Barr virus-associated tumors: Exosomes (Review). Oncol. Rep. 2022, 47, 13. [Google Scholar] [CrossRef] [PubMed]
- Morscio, J.; Dierickx, D.; Tousseyn, T. Molecular pathogenesis of B-cell posttransplant lymphoproliferative disorder: What do we know so far? Clin. Dev. Immunol. 2013, 2013, 150835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyauchi, K.; Urano, E.; Yoshiyama, H.; Komano, J. Cytokine signatures of transformed B cells with distinct Epstein-Barr virus latencies as a potential diagnostic tool for B cell lymphoma. Cancer Sci. 2011, 102, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Vaysberg, M.; Lambert, S.L.; Krams, S.M.; Martinez, O.M. Activation of the JAK/STAT pathway in Epstein Barr virus+-associated posttransplant lymphoproliferative disease: Role of interferon-gamma. Am. J. Transplant. 2009, 9, 2292–2302. [Google Scholar] [CrossRef] [Green Version]
- Rowe, M.; Peng-Pilon, M.; Huen, D.S.; Hardy, R.; Croom-Carter, D.; Lundgren, E.; Rickinson, A.B. Upregulation of bcl-2 by the Epstein-Barr virus latent membrane protein LMP1: A B-cell-specific response that is delayed relative to NF-kappa B activation and to induction of cell surface markers. J. Virol. 1994, 68, 5602–5612. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, W.S.; Park, C. Epstein-Barr virus latent membrane protein-1 protects B-cell lymphoma from rituximab-induced apoptosis through miR-155-mediated Akt activation and up-regulation of Mcl-1. Leuk. Lymphoma 2012, 53, 1586–1591. [Google Scholar] [CrossRef]
- Incrocci, R.; McCormack, M.; Swanson-Mungerson, M. Epstein-Barr virus LMP2A increases IL-10 production in mitogen-stimulated primary B-cells and B-cell lymphomas. J. Gen. Virol. 2013, 94 (Pt 5), 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Campion, E.M.; Hakimjavadi, R.; Loughran, S.T.; Phelan, S.; Smith, S.M.; D’Souza, B.N.; Tierney, R.J.; Bell, A.I.; Cahill, P.A.; Walls, D. Repression of the proapoptotic cellular BIK/NBK gene by Epstein-Barr virus antagonizes transforming growth factor β1-induced B-cell apoptosis. J. Virol. 2014, 88, 5001–5013. [Google Scholar] [CrossRef] [Green Version]
- Hong, G.K.; Gulley, M.L.; Feng, W.H.; Delecluse, H.J.; Holley-Guthrie, E.; Kenney, S.C. Epstein-Barr virus lytic infection contributes to lymphoproliferative disease in a SCID mouse model. J. Virol. 2005, 79, 13993–14003. [Google Scholar] [CrossRef] [Green Version]
- Perera, S.M.; Thomas, J.A.; Burke, M.; Crawford, D.H. Analysis of the T-cell micro-environment in Epstein-Barr virus-related post-transplantation B lymphoproliferative disease. J. Pathol. 1998, 184, 177–184. [Google Scholar] [CrossRef]
- Cui, X.; Snapper, C.M. Epstein Barr Virus: Development of Vaccines and Immune Cell Therapy for EBV-Associated Diseases. Front. Immunol. 2021, 12, 734471. [Google Scholar] [CrossRef] [PubMed]
- Rooney, C.M.; Smith, C.A.; Ng, C.Y.; Loftin, S.; Li, C.; Krance, R.A.; Brenner, M.K.; Heslop, H.E. Use of gene-modified virus-specific T lymphocytes to control Epstein-Barr-virus-related lymphoproliferation. Lancet 1995, 345, 9–13. [Google Scholar] [CrossRef]
- Comoli, P.; Labirio, M.; Basso, S.; Baldanti, F.; Grossi, P.; Furione, M.; Viganò, M.; Fiocchi, R.; Rossi, G.; Ginevri, F.; et al. Infusion of autologous Epstein-Barr virus (EBV)-specific cytotoxic T cells for prevention of EBV-related lymphoproliferative disorder in solid organ transplant recipients with evidence of active virus replication. Blood 2002, 99, 2592–2598. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Gloghini, A.; Pinto, A.; De Filippi, R.; Carbone, A. The classical Hodgkin’s lymphoma microenvironment and its role in promoting tumour growth and immune escape. J. Pathol. 2010, 221, 248–263. [Google Scholar] [CrossRef]
- Küppers, R. The biology of Hodgkin’s lymphoma. Nat. Rev. Cancer 2009, 9, 15–27. [Google Scholar] [CrossRef]
- Skinnider, B.F.; Mak, T.W. The role of cytokines in classical Hodgkin lymphoma. Blood 2002, 99, 4283–4297. [Google Scholar] [CrossRef] [Green Version]
- Aldinucci, D.; Lorenzon, D.; Cattaruzza, L.; Pinto, A.; Gloghini, A.; Carbone, A.; Colombatti, A. Expression of CCR5 receptors on Reed-Sternberg cells and Hodgkin lymphoma cell lines: Involvement of CCL5/Rantes in tumor cell growth and microenvironmental interactions. Int. J. Cancer 2008, 122, 769–776. [Google Scholar] [CrossRef]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-Barr virus-associated lymphomas. Philos. Trans. R Soc. Lond B Biol. Sci. 2017, 372, 20160271. [Google Scholar] [CrossRef]
- Roemer, M.G.; Advani, R.H.; Ligon, A.H.; Natkunam, Y.; Redd, R.A.; Homer, H.; Connelly, C.F.; Sun, H.H.; Daadi, S.E.; Freeman, G.J.; et al. PD-L1 and PD-L2 Genetic Alterations Define Classical Hodgkin Lymphoma and Predict Outcome. J. Clin. Oncol. 2016, 34, 2690–2697. [Google Scholar] [CrossRef] [Green Version]
- Bollard, C.M.; Gottschalk, S.; Torrano, V.; Diouf, O.; Ku, S.; Hazrat, Y.; Carrum, G.; Ramos, C.; Fayad, L.; Shpall, E.J.; et al. Sustained complete responses in patients with lymphoma receiving autologous cytotoxic T lymphocytes targeting Epstein-Barr virus latent membrane proteins. J. Clin. Oncol. 2014, 32, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, D.; Takada, K. Role of EBERs in the pathogenesis of EBV infection. Adv. Cancer Res. 2010, 107, 119–136. [Google Scholar]
- Tsimbouri, P.; Al-Sheikh, Y.; Drotar, M.E.; Cushley, W.; Wilson, J.B. Epstein-Barr virus nuclear antigen-1 renders lymphocytes responsive to IL-2 but not IL-15 for survival. J. Gen. Virol. 2008, 89 (Pt 11), 2821–2832. [Google Scholar] [CrossRef] [PubMed]
- Granai, M.; Mundo, L.; Akarca, A.U.; Siciliano, M.C.; Rizvi, H.; Mancini, V.; Onyango, N.; Nyagol, J.; Abinya, N.O.; Maha, I.; et al. Immune landscape in Burkitt lymphoma reveals M2-macrophage polarization and correlation between PD-L1 expression and non-canonical EBV latency program. Infect. Agents Cancer 2020, 15, 28. [Google Scholar] [CrossRef] [PubMed]
- Crombie, J.; LaCasce, A. The treatment of Burkitt lymphoma in adults. Blood 2021, 137, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Que, Y.; Wang, J.; Zhu, J.; Li, N.; Huang, J.; Lu, S.; Sun, F.; Zhang, L.; Zhen, Z.; Zhang, L.; et al. Combination Therapy With Anti-PD-1 or PD-1 Antibody Alone in Asian Pediatric Patients With Relapsed or Refractory Cancer. Front. Immunol. 2021, 12, 647733. [Google Scholar] [CrossRef]
- Heslop, H.E. Biology and treatment of Epstein-Barr virus-associated non-Hodgkin lymphomas. Hematol. Am. Soc. Hematol. Educ. Program 2005, 260–266. [Google Scholar] [CrossRef] [Green Version]
- Ciavarella, S.; Vegliante, M.C.; Fabbri, M.; De Summa, S.; Melle, F.; Motta, G.; De Iuliis, V.; Opinto, G.; Enjuanes, A.; Rega, S.; et al. Dissection of DLBCL microenvironment provides a gene expression-based predictor of survival applicabl e to formalin-fixed paraffin-embedded tissue. Ann. Oncol. 2018, 29, 2363–2370. [Google Scholar] [CrossRef] [Green Version]
- Xie, Y.; Pittaluga, S.; Jaffe, E.S. The histological classification of diffuse large B-cell lymphomas. Semin. Hematol. 2015, 52, 57–66. [Google Scholar] [CrossRef] [Green Version]
- Kanegane, H.; Nomura, K.; Miyawaki, T.; Tosato, G. Biological aspects of Epstein-Barr virus (EBV)-infected lymphocytes in chronic active EBV infection and associated malignancies. Crit. Rev. Oncol. Hematol. 2002, 44, 239–249. [Google Scholar] [CrossRef]
- Quan, L.; Chen, X.; Liu, A.; Zhang, Y.; Guo, X.; Yan, S.; Liu, Y. PD-1 Blockade Can Restore Functions of T-Cells in Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma In Vitro. PLoS ONE 2015, 10, e0136476. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.; Vistarop, A.G.; Huaman, F.; Narbaitz, M.; Metrebian, F.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Cytotoxic response against Epstein Barr virus coexists with diffuse large B-cell lymphoma tolerogenic microenvironment: Clinical features and survival impact. Sci. Rep. 2017, 7, 10813. [Google Scholar] [CrossRef] [PubMed]
- Cárdenas, D.; Vélez, G.; Orfao, A.; Herrera, M.V.; Solano, J.; Olaya, M.; Uribe, A.M.; Saavedra, C.; Duarte, M.; Rodríguez, M.; et al. Epstein-Barr virus-specific CD8(+) T lymphocytes from diffuse large B cell lymphoma patients are functionally impaired. Clin. Exp. Immunol. 2015, 182, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keane, C.; Tobin, J.; Gunawardana, J.; Francis, S.; Gifford, G.; Gabrielli, S.; Gill, A.; Stevenson, W.; Talaulikar, D.; Gould, C.; et al. The tumour microenvironment is immuno-tolerogenic and a principal determinant of patient outcome in E BV-positive diffuse large B-cell lymphoma. Eur. J. Haematol. 2019, 103, 200–207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondello, P.; Mian, M. Frontline treatment of diffuse large B-cell lymphoma: Beyond R-CHOP. Hematol. Oncol. 2019, 37, 333–344. [Google Scholar] [CrossRef]
- Chavez, J.C.; Locke, F.L. CAR T cell therapy for B-cell lymphomas. Best Pract. Research. Clin. Haematol. 2018, 31, 135–146. [Google Scholar] [CrossRef]
- Cherkassky, L.; Morello, A.; Villena-Vargas, J.; Feng, Y.; Dimitrov, D.S.; Jones, D.R.; Sadelain, M.; Adusumilli, P.S. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Investig. 2016, 126, 3130–3144. [Google Scholar] [CrossRef] [Green Version]
- Takahara, M.; Kis, L.L.; Nagy, N.; Liu, A.; Harabuchi, Y.; Klein, G.; Klein, E. Concomitant increase of LMP1 and CD25 (IL-2-receptor alpha) expression induced by IL-10 in the EBV-positive NK lines SNK6 and KAI3. Int. J. Cancer 2006, 119, 2775–2783. [Google Scholar] [CrossRef]
- Yoshino, K.; Kishibe, K.; Nagato, T.; Ueda, S.; Komabayashi, Y.; Takahara, M.; Harabuchi, Y. Expression of CD70 in nasal natural killer/T cell lymphoma cell lines and patients; its role for cell proliferation through binding to soluble CD27. Br. J. Haematol. 2013, 160, 331–342. [Google Scholar] [CrossRef]
- Chuang, H.C.; Lay, J.D.; Hsieh, W.C.; Su, I.J. Pathogenesis and mechanism of disease progression from hemophagocytic lymphohistiocytosis to Epstein- Barr virus-associated T-cell lymphoma: Nuclear factor-kappa B pathway as a potential therapeutic target. Cancer Sci. 2007, 98, 1281–1287. [Google Scholar] [CrossRef]
- Bi, X.W.; Wang, H.; Zhang, W.W.; Wang, J.H.; Liu, W.J.; Xia, Z.J.; Huang, H.Q.; Jiang, W.Q.; Zhang, Y.J.; Wang, L. PD-L1 is upregulated by EBV-driven LMP1 through NF-κB pathway and correlates with poor prognosis in natural killer/T-cell lymphoma. J. Hematol. Oncol. 2016, 9, 109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, E.; Kwong, Y.L. The diagnosis and management of NK/T-cell lymphomas. J. Hematol. Oncol. 2017, 10, 85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwong, Y.L.; Chan, T.S.Y.; Tan, D.; Kim, S.J.; Poon, L.M.; Mow, B.; Khong, P.L.; Loong, F.; Au-Yeung, R.; Iqbal, J.; et al. PD1 blockade with pembrolizumab is highly effective in relapsed or refractory NK/T-cell lymphoma failing l-asparaginase. Blood 2017, 129, 2437–2442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, E.T.; Adami, H.O. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1765–1777. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.P.; Chan, A.T.C.; Le, Q.T.; Blanchard, P.; Sun, Y.; Ma, J. Nasopharyngeal carcinoma. Lancet 2019, 394, 64–80. [Google Scholar] [CrossRef]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Raab-Traub, N. Nasopharyngeal Carcinoma: An Evolving Role for the Epstein-Barr Virus. Curr. Top. Microbiol. Immunol. 2015, 390 (Pt 1), 339–363. [Google Scholar]
- Lin, D.C.; Meng, X.; Hazawa, M.; Nagata, Y.; Varela, A.M.; Xu, L.; Sato, Y.; Liu, L.Z.; Ding, L.W.; Sharma, A.; et al. The genomic landscape of nasopharyngeal carcinoma. Nat. Genet. 2014, 46, 866–871. [Google Scholar] [CrossRef]
- Lau, K.M.; Cheng, S.H.; Lo, K.W.; Lee, S.A.; Woo, J.K.; van Hasselt, C.A.; Lee, S.P.; Rickinson, A.B.; Ng, M.H. Increase in circulating Foxp3+CD4+CD25(high) regulatory T cells in nasopharyngeal carcinoma patients. Br. J. Cancer 2007, 96, 617–622. [Google Scholar] [CrossRef] [Green Version]
- Mai, H.Q.; Chen, Q.Y.; Chen, D.; Hu, C.; Yang, K.; Wen, J.; Li, J.; Shi, Y.R.; Jin, F.; Xu, R.; et al. Toripalimab or placebo plus chemotherapy as first-line treatment in advanced nasopharyngeal carcinoma: A multicenter randomized phase 3 trial. Nat. Med. 2021, 27, 1536–1543. [Google Scholar] [CrossRef]
- Li, F.; Song, D.; Lu, Y.; Zhu, H.; Chen, Z.; He, X. Delayed-type hypersensitivity (DTH) immune response related with EBV-DNA in nasopharyngeal carcinoma treated with autologous dendritic cell vaccination after radiotherapy. J. Immunother. 2013, 36, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Hui, E.P.; Taylor, G.S.; Jia, H.; Ma, B.B.; Chan, S.L.; Ho, R.; Wong, W.L.; Wilson, S.; Johnson, B.F.; Edwards, C.; et al. Phase I trial of recombinant modified vaccinia ankara encoding Epstein-Barr viral tumor antigens in nasopharyngeal carcinoma patients. Cancer Res. 2013, 73, 1676–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasari, V.; Sinha, D.; Neller, M.A.; Smith, C.; Khanna, R. Prophylactic and therapeutic strategies for Epstein-Barr virus-associated diseases: Emerging strategies for clinical development. Expert Rev. Vaccines 2019, 18, 457–474. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Lee, J.; Nebozhyn, M.; Kim, K.M.; Ting, J.C.; Wong, S.S.; Liu, J.; Yue, Y.G.; Wang, J.; Yu, K.; et al. Molecular analysis of gastric cancer identifies subtypes associated with distinct clinical outcomes. Nat. Med. 2015, 21, 449–456. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, S.H.; Han, S.H.; An, J.S.; Lee, E.S.; Kim, Y.S. Clinicopathological and molecular characteristics of Epstein-Barr virus-associated gastric carcinoma: A meta-analysis. J. Gastroenterol. Hepatol. 2009, 24, 354–365. [Google Scholar] [CrossRef]
- Fukayama, M.; Hino, R.; Uozaki, H. Epstein-Barr virus and gastric carcinoma: Virus-host interactions leading to carcinoma. Cancer Sci. 2008, 99, 1726–1733. [Google Scholar] [CrossRef]
- Fukayama, M.; Ushiku, T. Epstein-Barr virus-associated gastric carcinoma. Pathol. Res. Pract. 2011, 207, 529–537. [Google Scholar] [CrossRef]
- Fukayama, M.; Abe, H.; Kunita, A.; Shinozaki-Ushiku, A.; Matsusaka, K.; Ushiku, T.; Kaneda, A. Thirty years of Epstein-Barr virus-associated gastric carcinoma. Virchows Arch. 2020, 476, 353–365. [Google Scholar] [CrossRef]
- Fukayama, M.; Kunita, A.; Kaneda, A. Gastritis-Infection-Cancer Sequence of Epstein-Barr Virus-Associated Gastric Cancer. Adv. Exp. Med. Biol. 2018, 1045, 437–457. [Google Scholar]
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef] [Green Version]
- Ribeiro, J.; Oliveira, C.; Malta, M.; Sousa, H. Epstein-Barr virus gene expression and latency pattern in gastric carcinomas: A systematic review. Future Oncol. 2017, 13, 567–579. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Park, C.; Kim, H.J.; Park, J.; Hwang, J.; Kim, J.I.; Choi, M.G.; Kim, S.; Kim, K.M.; Kang, M.S. Deregulation of immune response genes in patients with Epstein-Barr virus-associated gastric cancer and outcomes. Gastroenterology 2015, 148, 137–147.e9. [Google Scholar] [CrossRef] [PubMed]
- Song, H.J.; Srivastava, A.; Lee, J.; Kim, Y.S.; Kim, K.M.; Ki Kang, W.; Kim, M.; Kim, S.; Park, C.K.; Kim, S. Host inflammatory response predicts survival of patients with Epstein-Barr virus-associated gastric carcinoma. Gastroenterology 2010, 139, 84–92.e2. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.; Abe, H.; Kunita, A.; Yamashita, H.; Seto, Y.; Fukayama, M. Overexpression and gene amplification of PD-L1 in cancer cells and PD-L1+ immune cells in Epstein-Barr virus-associated gastric cancer: The prognostic implications. Mod. Pathol. 2017, 30, 427–439. [Google Scholar] [CrossRef]
- Zhang, N.N.; Chen, J.N.; Xiao, L.; Tang, F.; Zhang, Z.G.; Zhang, Y.W.; Feng, Z.Y.; Jiang, Y.; Shao, C.K. Accumulation Mechanisms of CD4(+)CD25(+)FOXP3(+) Regulatory T Cells in EBV-associated Gastric Carcinoma. Sci. Rep. 2015, 5, 18057. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, T.; Abe, H.; Morikawa, T.; Yamashita, H.; Ishikawa, S.; Ushiku, T.; Seto, Y.; Fukayama, M. Low density of CD204-positive M2-type tumor-associated macrophages in Epstein-Barr virus-associated gastric cancer: A clinicopathologic study with digital image analysis. Hum. Pathol. 2016, 56, 74–80. [Google Scholar] [CrossRef]
- Strong, M.J.; Xu, G.; Coco, J.; Baribault, C.; Vinay, D.S.; Lacey, M.R.; Strong, A.L.; Lehman, T.A.; Seddon, M.B.; Lin, Z.; et al. Differences in gastric carcinoma microenvironment stratify according to EBV infection intensity: Implications for possible immune adjuvant therapy. PLoS Pathog. 2013, 9, e1003341. [Google Scholar] [CrossRef]
- Naseem, M.; Barzi, A.; Brezden-Masley, C.; Puccini, A.; Berger, M.D.; Tokunaga, R.; Battaglin, F.; Soni, S.; McSkane, M.; Zhang, W.; et al. Outlooks on Epstein-Barr virus associated gastric cancer. Cancer Treat. Rev. 2018, 66, 15–22. [Google Scholar] [CrossRef]
- Huang, Y.H.; Zhang, C.Z.; Huang, Q.S.; Yeong, J.; Wang, F.; Yang, X.; He, Y.F.; Zhang, X.L.; Zhang, H.; Chen, S.L.; et al. Clinicopathologic features, tumor immune microenvironment and genomic landscape of Epstein-Barr virus -associated intrahepatic cholangiocarcinoma. J. Hepatol. 2021, 74, 838–849. [Google Scholar] [CrossRef]
- Hussein, K.; Maecker-Kolhoff, B.; Donnerstag, F.; Laenger, F.; Kreipe, H.; Jonigk, D. Epstein-Barr virus-associated smooth muscle tumours after transplantation, infection with human immunodeficiency virus and congenital immunodeficiency syndromes. Pathobiology 2013, 80, 297–301. [Google Scholar] [CrossRef]
- Lee, E.S.; Locker, J.; Nalesnik, M.; Reyes, J.; Jaffe, R.; Alashari, M.; Nour, B.; Tzakis, A.; Dickman, P.S. The association of Epstein-Barr virus with smooth-muscle tumors occurring after organ transplantation. N. Engl. J. Med. 1995, 332, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Calafiore, R.; Mouchtouris, N.; Flomenberg, N.; Harrop, J.S. Epstein-Barr Virus-Associated Smooth Muscle Tumor of the Spine After Bone Marrow Transplant: Case Report and Review of Literature. World Neurosurg. 2020, 135, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Hussein, K.; Rath, B.; Ludewig, B.; Kreipe, H.; Jonigk, D. Clinico-pathological characteristics of different types of immunodeficiency-associated smooth muscle tumours. Eur. J. Cancer 2014, 50, 2417–2424. [Google Scholar] [CrossRef] [PubMed]
- Rogatsch, H.; Bonatti, H.; Menet, A.; Larcher, C.; Feichtinger, H.; Dirnhofer, S. Epstein-Barr virus-associated multicentric leiomyosarcoma in an adult patient after heart transplantation: Case report and review of the literature. Am. J. Surg. Pathol. 2000, 24, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Deyrup, A.T.; Lee, V.K.; Hill, C.E.; Cheuk, W.; Toh, H.C.; Kesavan, S.; Chan, E.W.; Weiss, S.W. Epstein-Barr virus-associated smooth muscle tumors are distinctive mesenchymal tumors reflecting multiple infection events: A clinicopathologic and molecular analysis of 29 tumors from 19 patients. Am. J. Surg. Pathol. 2006, 30, 75–82. [Google Scholar] [CrossRef]
- Creager, A.J.; Maia, D.M.; Funkhouser, W.K. Epstein-Barr virus-associated renal smooth muscle neoplasm: Report of a case with review of the literature. Arch. Pathol. Lab. Med. 1998, 122, 277–281. [Google Scholar]
- Chong, Y.B.; Lu, P.-L.; Ma, Y.-C.; Yin, H.-L.; Chang, C.-H. Epstein-Barr Virus-Associated Smooth Muscle Tumor and Its Correlation With CD4 Levels in a Patient With HIV Infection. Front. Cell. Infect. Microbiol. 2022, 12, 725342. [Google Scholar] [CrossRef]
- Chen, B.J.; Chapuy, B.; Ouyang, J.; Sun, H.H.; Roemer, M.G.; Xu, M.L.; Yu, H.; Fletcher, C.D.; Freeman, G.J.; Shipp, M.A.; et al. PD-L1 expression is characteristic of a subset of aggressive B-cell lymphomas and virus-associated malignancies. Clin. Cancer Res. 2013, 19, 3462–3473. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Masterson, L.; Howard, J.; Gonzalez-Cruz, J.; Jackson, C.; Barnett, C.; Overton, L.; Liu, H.; Ladwa, R.; Simpson, F.; McGrath, M.; et al. Immune checkpoint inhibitors in advanced nasopharyngeal carcinoma: Beyond an era of chemoradiation? Int. J. Cancer 2020, 146, 2305–2314. [Google Scholar] [CrossRef]
- Burns, D.M.; Crawford, D.H. Epstein-Barr virus-specific cytotoxic T-lymphocytes for adoptive immunotherapy of post-transplant lymphoproliferative disease. Blood Rev. 2004, 18, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Burton, R.; Reddy, V.; Lucas, K.G. Safety of allogeneic Epstein-Barr virus (EBV)-specific cytotoxic T lymphocytes for patients with refractory EBV-related lymphoma. Br. J. Haematol. 2002, 118, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Poppema, S. Immunobiology and pathophysiology of Hodgkin lymphomas. Hematol. Am. Soc. Hematol. Educ. Program 2005, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Duraiswamy, J.; Sherritt, M.; Thomson, S.; Tellam, J.; Cooper, L.; Connolly, G.; Bharadwaj, M.; Khanna, R. Therapeutic LMP1 polyepitope vaccine for EBV-associated Hodgkin disease and nasopharyngeal carcinoma. Blood 2003, 101, 3150–3156. [Google Scholar] [CrossRef] [Green Version]
- Münz, C.; Bickham, K.L.; Subklewe, M.; Tsang, M.L.; Chahroudi, A.; Kurilla, M.G.; Zhang, D.; O’Donnell, M.; Steinman, R.M. Human CD4(+) T lymphocytes consistently respond to the latent Epstein-Barr virus nuclear antigen EBNA 1. J. Exp. Med. 2000, 191, 1649–1660. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.P.; Tierney, R.J.; Thomas, W.A.; Brooks, J.M.; Rickinson, A.B. Conserved CTL epitopes within EBV latent membrane protein 2: A potential target for CTL-based tumor therapy. J. Immunol. 1997, 158, 3325–3334. [Google Scholar]
- Lin, C.L.; Lo, W.F.; Lee, T.H.; Ren, Y.; Hwang, S.L.; Cheng, Y.F.; Chen, C.L.; Chang, Y.S.; Lee, S.P.; Rickinson, A.B.; et al. Immunization with Epstein-Barr Virus (EBV) peptide-pulsed dendritic cells induces functional CD8+ T-c ell immunity and may lead to tumor regression in patients with EBV-positive nasopharyngeal carcinoma. Cancer Res. 2002, 62, 6952–6958. [Google Scholar]
- Si, Y.; Deng, Z.; Lan, G.; Du, H.; Wang, Y.; Si, J.; Wei, J.; Weng, J.; Qin, Y.; Huang, B.; et al. The Safety and Immunological Effects of rAd5-EBV-LMP2 Vaccine in Nasopharyngeal Carcinoma Patients: A Phase I Clinical Trial and Two-Year Follow-Up. Chem. Pharm. Bull. 2016, 64, 1118–1123. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| EBV-Associated Malignancies | EBV Positive Rate | Latency Pattern | Immune Markers | Immunotherapy | |
|---|---|---|---|---|---|
| Immune Cells | Immune Molecules | ||||
| PTLD | 100% | Latency III | Memory/helper T cells Decreased cytotoxic T cells | IFN-γ, IL-6, IL-10, IL-13 | Adoptive T cell therapy |
| Classical HL | 50% | Latency II | Tregs Exhausted CD8+ T cells | PD-L1, TNFR, Th2 cytokines and chemokines, IL-10, galectin-1, TGF-β | PD-1 inhibitors T cell therapy |
| Epidemic BL | 100% | Latency I | M2 TAMs | IL-2, IL-6, IL-10 | NA |
| DLBCL | 10% | Latency II or III | M2 TAMs Exhausted CD8+ T cells | IL-10, PD-1, PD-L1, PD-L2, LAG3, TIM3 | Chimeric antigen receptor T cell therapy |
| Extranodal NK/T cell lymphomas | 100% | Latency II | Activated T cells and macrophages | IL-2, IL-10, CD27, TNF-α, PD-L1 | PD-1 inhibitors |
| Undifferentiated NPC | 100% | Latency I/II | Exhausted CD8+ T cells LAMP3+ DCs | PD-L1, PD-L2, CTLA-4, IDO1, HLA-G | PD-1 inhibitors Therapeutic EBV vaccines |
| GC | 10% | Latency I or I/II | CD8+ T cells Tregs TAMs | PD-L1, IDO1 | PD-1 inhibitors |
| ICC | 6.6% | Latency I | CD8+ T cells M1 TAMs CD20+ B cells | PD-L1 | NA |
| SMT | <1% | Latency III | T cells | NA | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zheng, X.; Huang, Y.; Li, K.; Luo, R.; Cai, M.; Yun, J. Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein–Barr Virus-Associated Malignancies. Viruses 2022, 14, 1017. https://doi.org/10.3390/v14051017
Zheng X, Huang Y, Li K, Luo R, Cai M, Yun J. Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein–Barr Virus-Associated Malignancies. Viruses. 2022; 14(5):1017. https://doi.org/10.3390/v14051017
Chicago/Turabian StyleZheng, Xueyi, Yuhua Huang, Kai Li, Rongzhen Luo, Muyan Cai, and Jingping Yun. 2022. "Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein–Barr Virus-Associated Malignancies" Viruses 14, no. 5: 1017. https://doi.org/10.3390/v14051017
APA StyleZheng, X., Huang, Y., Li, K., Luo, R., Cai, M., & Yun, J. (2022). Immunosuppressive Tumor Microenvironment and Immunotherapy of Epstein–Barr Virus-Associated Malignancies. Viruses, 14(5), 1017. https://doi.org/10.3390/v14051017