
Jingmen Tick Virus in Ticks from Kenya

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Material and Methods
2.1. Ethical Approval
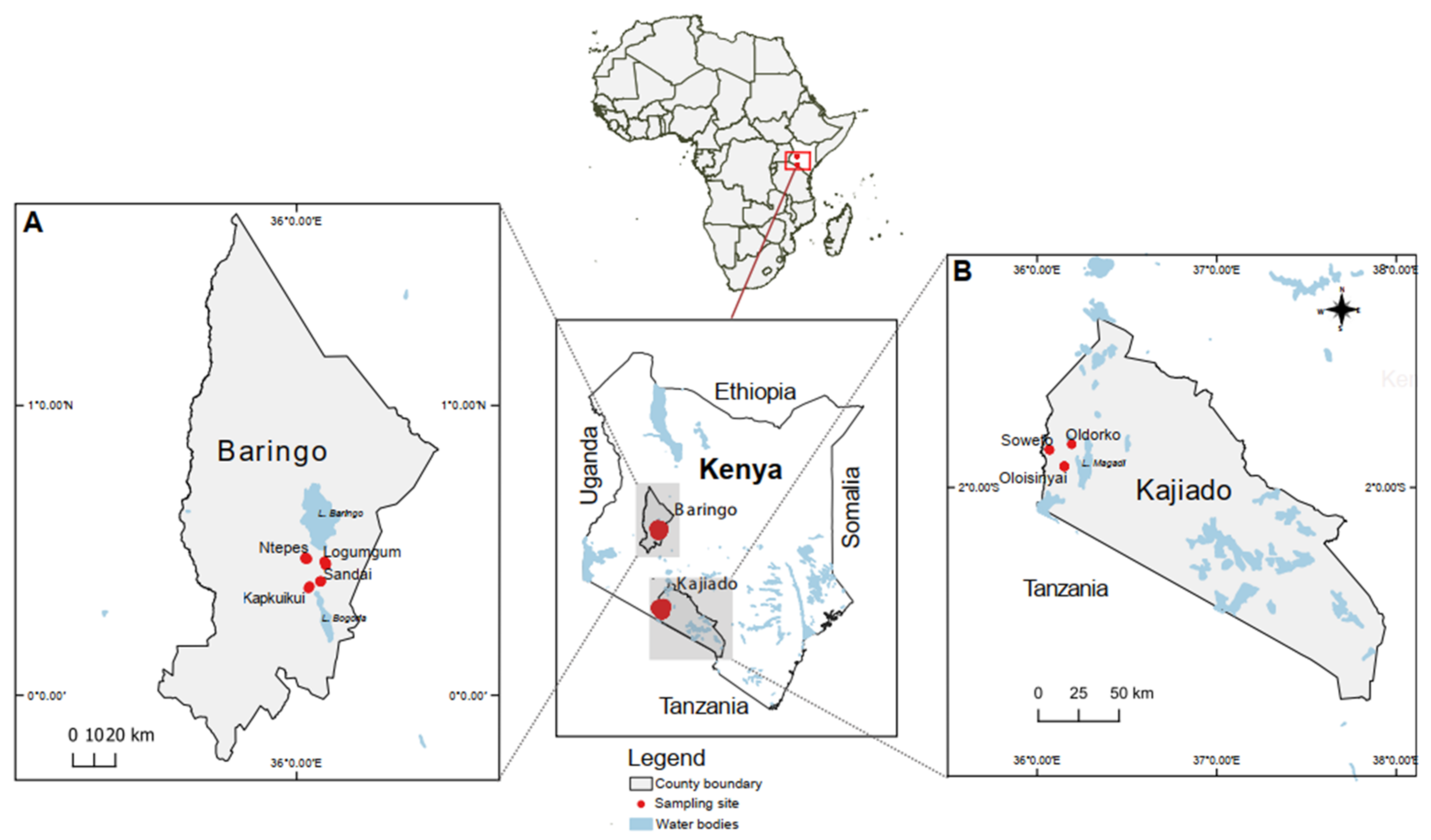
2.2. Study Sites, Tick Sampling and Morphological Identification
2.3. Tortoise Sampling
2.4. Tick Homogenization and Molecular Identification
2.5. RNA Extraction and PCR Screening
2.6. Virus Isolation and Quantification of Viral Genome Copies
2.7. Library Preparation and Next-Generation Sequencing
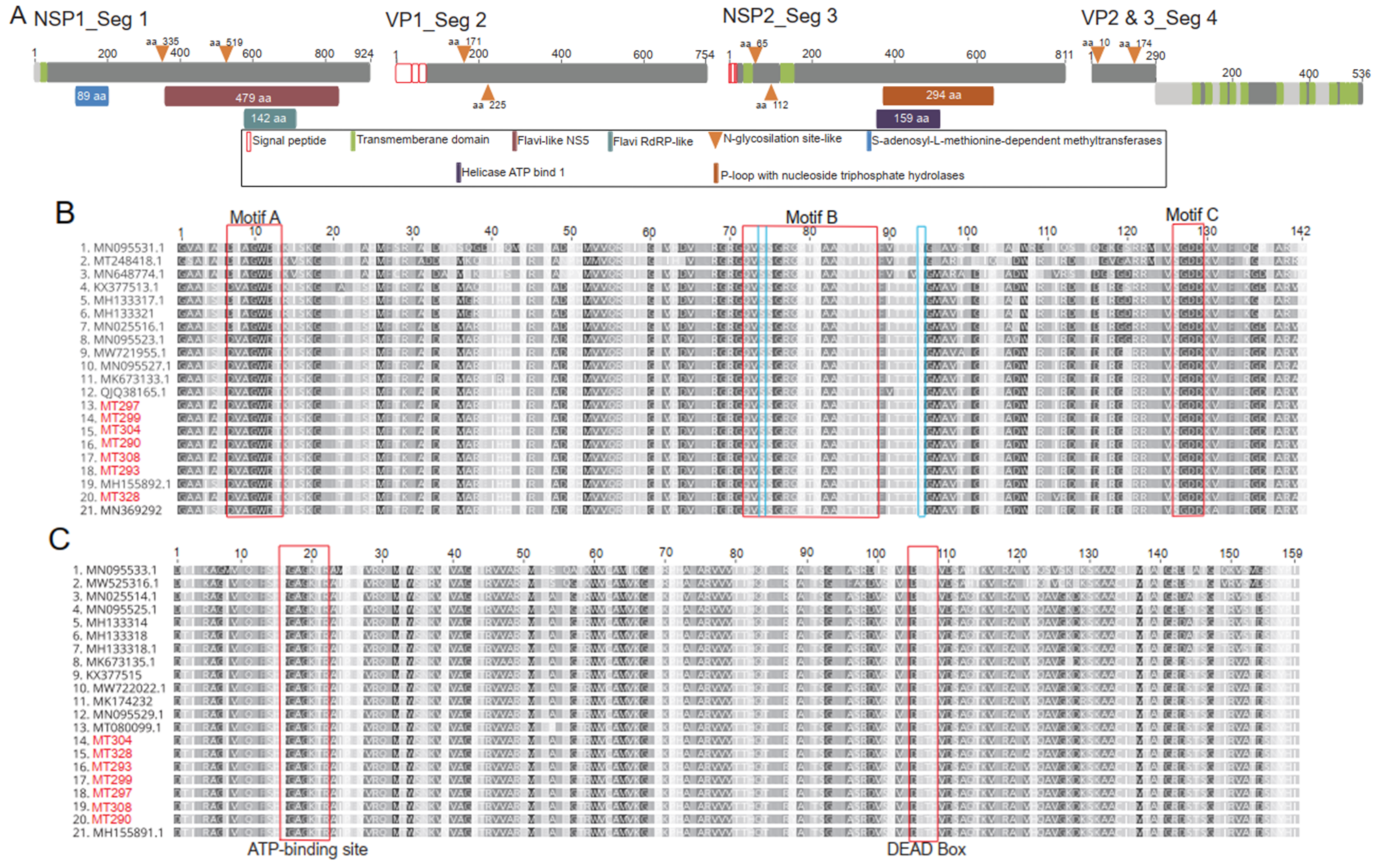
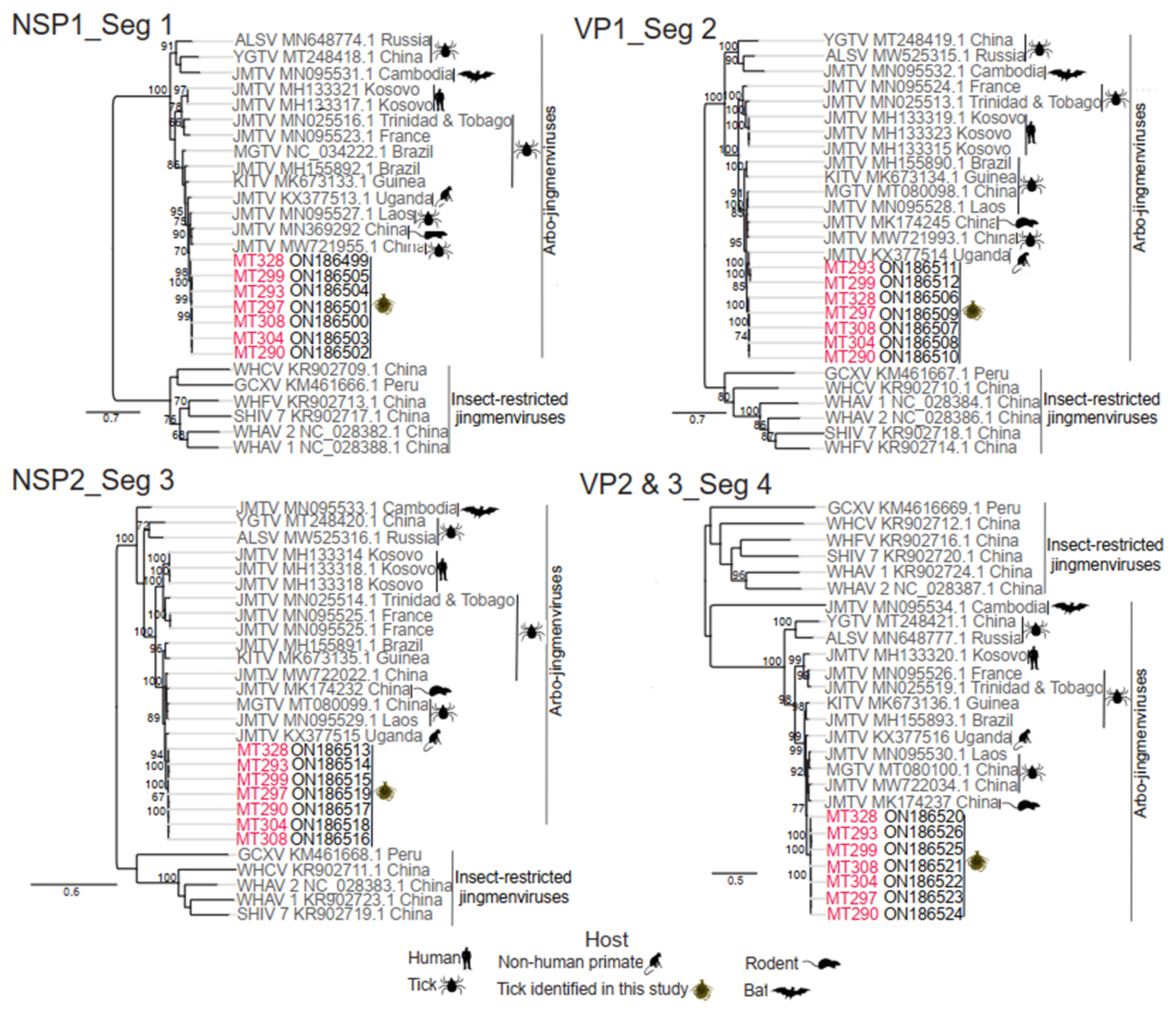
2.8. Phylogenetic Analysis and Genome Characterization
2.9. Statistical Analysis
2.10. Sequence Accession Numbers
3. Results
3.1. Tick Collection from Livestock and Tortoises
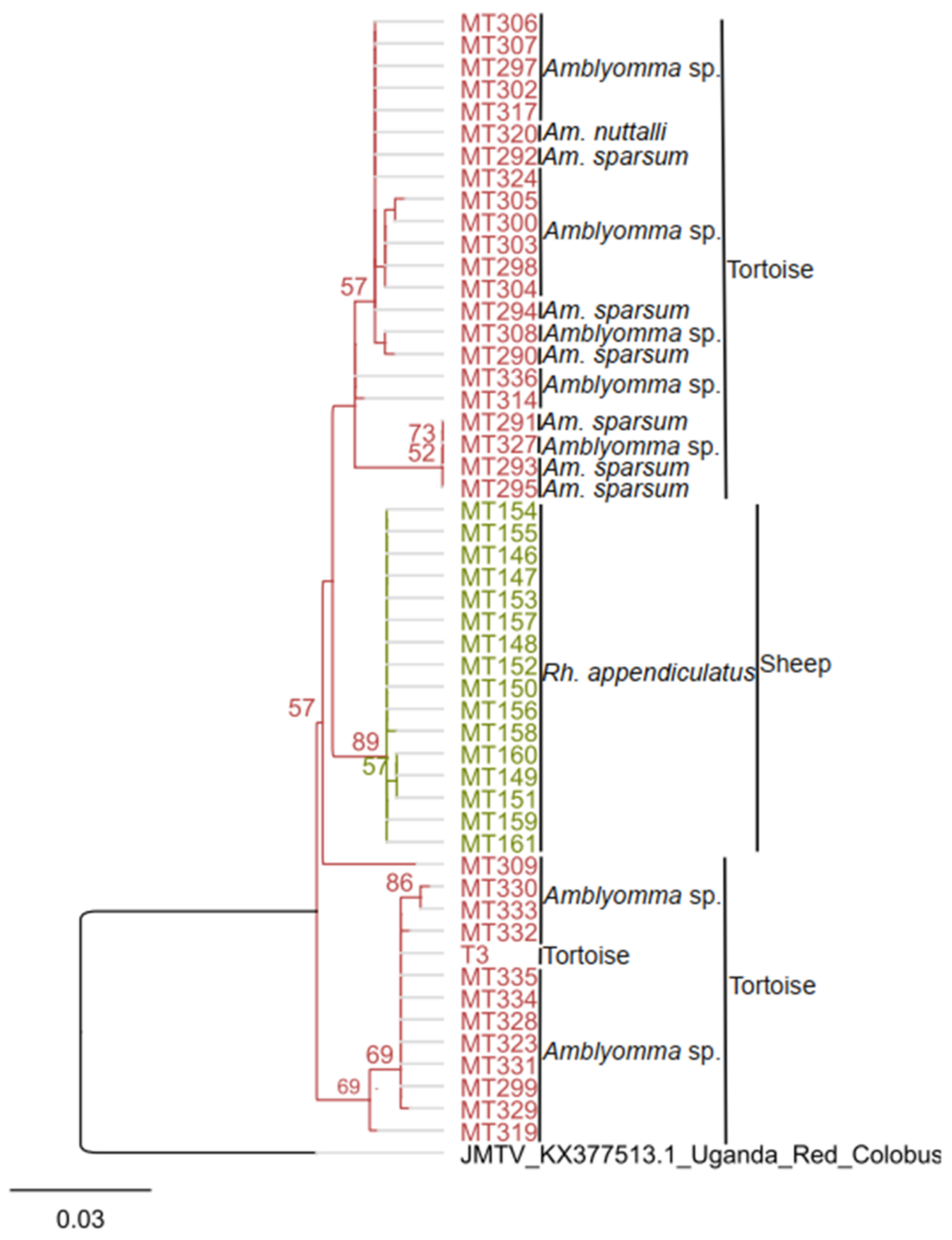
3.2. JMTV Infection in Ticks and Tortoises
3.3. Virus Isolation and Genome Organization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclosure Statement
References
- Qin, X.C.; Shi, M.; Tian, J.H.; Lin, X.D.; Gao, D.Y.; He, J.R.; Wang, J.B.; Li, C.X.; Kang, Y.J.; Yu, B.; et al. A Tick-Borne Segmented RNA Virus Contains Genome Segments Derived from Unsegmented Viral Ancestors. Proc. Natl. Acad. Sci. USA 2014, 111, 6744–6749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, N.; Liu, H.B.; Ni, X.B.; Bell-Sakyi, L.; Zheng, Y.C.; Song, J.L.; Li, J.; Jiang, B.G.; Wang, Q.; Sun, Y.; et al. Emergence of Human Infection with Jingmen Tick Virus in China: A Retrospective Study. eBioMedicine 2019, 43, 317–324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmerich, P.; Jakupi, X.; von Possel, R.; Berisha, L.; Halili, B.; Günther, S.; Cadar, D.; Ahmeti, S.; Schmidt-Chanasit, J. Viral Metagenomics, Genetic and Evolutionary Characteristics of Crimean-Congo Hemorrhagic Fever Orthonairovirus in Humans, Kosovo. Infect. Genet. Evol. 2018, 65, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Ladner, J.T.; Wiley, M.R.; Beitzel, B.; Auguste, A.J.; Dupuis, A.; Lindquist, M.E.; Sibley, S.D.; Kota, K.P.; Fetterer, D.; Eastwood, G.; et al. A Multicomponent Animal Virus Isolated from Mosquitoes. Physiol. Behav. 2017, 176, 139–148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuivanen, S.; Levanov, L.; Kareinen, L.; Sironen, T.; Jääskeläinen, A.J.; Plyusnin, I.; Zakham, F.; Emmerich, P.; Schmidt-Chanasit, J.; Hepojoki, J.; et al. Detection of Novel Tick-Borne Pathogen, Alongshan Virus, in Ixodes Ricinus Ticks, South-Eastern Finland, 2019. Eurosurveillance 2019, 24, 1900394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kholodilov, I.S.; Litov, A.G.; Klimentov, A.S.; Belova, O.A.; Polienko, A.E.; Nikitin, N.A.; Shchetinin, A.M.; Ivannikova, A.Y.; Bell-Sakyi, L.; Yakovlev, A.S.; et al. Isolation and Characterisation of Alongshan Virus in Russia. Viruses 2020, 12, 362. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.D.; Wang, B.; Wei, F.; Han, S.Z.; Zhang, L.; Yang, Z.T.; Yan, Y.; Lv, X.L.; Li, L.; Wang, S.C.; et al. A New Segmented Virus Associated with Human Febrile Illness in China. N. Engl. J. Med. 2019, 380, 2116–2125. [Google Scholar] [CrossRef]
- Guo, J.J.; Lin, X.D.; Chen, Y.M.; Hao, Z.Y.; Wang, Z.X.; Yu, Z.M.; Lu, M.; Li, K.; Qin, X.C.; Wang, W.; et al. Diversity and Circulation of Jingmen Tick Virus in Ticks and Mammals. Virus Evol. 2020, 6, veaa051. [Google Scholar] [CrossRef]
- Yu, Z.M.; Chen, J.T.; Qin, J.; Guo, J.J.; Li, K.; Xu, Q.Y.; Wang, W.; Lu, M.; Qin, X.C.; Zhang, Y.Z. Identification and Characterization of Jingmen Tick Virus in Rodents from Xinjiang, China. Infect. Genet. Evol. 2020, 84, 104411. [Google Scholar] [CrossRef]
- Shi, M.; Lin, X.-D.; Vasilakis, N.; Tian, J.-H.; Li, C.-X.; Chen, L.-J.; Eastwood, G.; Diao, X.-N.; Chen, M.-H.; Chen, X.; et al. Divergent Viruses Discovered in Arthropods and Vertebrates Revise the Evolutionary History of the Flaviviridae and Related Viruses. J. Virol. 2016, 90, 659–669. [Google Scholar] [CrossRef] [Green Version]
- Temmam, S.; Bigot, T.; Chrétien, D.; Gondard, M.; Pérot, P.; Pommelet, V. Insights into the Host Range, Genetic Diversity, and Geographical Distribution of Jingmenviruses. Ecol. Evol. Sci. 2019, 4, e00645-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garry, C.E.; Garry, R.F. Proteomics Computational Analyses Suggest That the Envelope Glycoproteins of Segmented Jingmen Flavi-like Viruses Are Class II Viral Fusion Proteins (β-Penetrenes) with Mucin-like Domains. Viruses 2020, 12, 260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tigoi, C.; Lwande, O.; Orindi, B.; Irura, Z.; Ongus, J.; Sang, R. Seroepidemiology of Selected Arboviruses in Febrile Patients Visiting Selected Health Facilities in the Lake/River Basin Areas of Lake Baringo, Lake Naivasha, and Tana River, Kenya. Vector-Borne Zoonotic Dis. 2015, 15, 124–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ajamma, Y.U.; Onchuru, T.O.; Ouso, D.O.; Omondi, D.; Masiga, D.K.; Villinger, J. Vertical Transmission of Naturally Occurring Bunyamwera and Insect-Specific Flavivirus Infections in Mosquitoes from Islands and Mainland Shores of Lakes Victoria and Baringo in Kenya. PLoS Negl. Trop. Dis. 2018, 12, e0006949. [Google Scholar] [CrossRef] [PubMed]
- Tchouassi, D.P.; Marklewitz, M.; Chepkorir, E.; Zirkel, F.; Agha, S.B.; Tigoi, C.C.; Koskei, E.; Drosten, C.; Borgemeister, C.; Torto, B.; et al. Sandfly-Associated Phlebovirus with Evidence of Neutralizing Antibodies in Humans, Kenya. Emerg. Infect. Dis. 2019, 25, 681–690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marklewitz, M.; Tchouassi, D.P.; Hieke, C.; Heyde, V.; Torto, B.; Sang, R.; Junglen, S. Insights into the Evolutionary Origin of Mediterranean Sandfly Fever Viruses. mSphere 2020, 5, e00598-20. [Google Scholar] [CrossRef]
- QGIS Development Team. Welcome to the QGIS Project! Available online: https://qgis.org/en/site/ (accessed on 11 June 2019).
- Harrison, B.A.; Whitt, P.B.; Roberts, L.F.; Lehman, J.A.; Lindsey, N.P.; Nasci, R.S.; Hansen, G.R. Rapid Assessment of Mosquitoes and Arbovirus Activity after Floods in Southeastern Kansas, 2007. J. Am. Mosq. Control Assoc. 2009, 25, 265–271. [Google Scholar] [CrossRef]
- John, G.; Matthysse, J.G.M. The Ixodid Ticks of Uganda Together with Species Pertinent to Uganda Because of Their Present Known Distribution; Entomological Society of America: Annapolis, MA, USA, 1987. [Google Scholar]
- Okello-Onen, J.; Hassan, S.M.; Essuman, S. Taxonomy of African Ticks: An Identification Manual, 1st ed.; International Centre of Insect Physiology and Ecology, African Postgraduate Programme in Insect Science: Nairobi, Kenya, 1999. [Google Scholar]
- Walker, J.; Keirans, J.E.; Horak, I.G. Rhipicephalud (Acari, Ixodidae)—A Guide to Brown Ticks of the World; Cambridge University Press: Cambridge, UK, 2000. [Google Scholar]
- Walker, A.R.; Bouattour, A.; Camicas, J.-L.; Estrada-Peña, A.; Horak, I.G.; Latif, A.A.; Pegram, R.G.; Preston, P.M. Ticks of Domestic Animals in Municipal Abattoir for Their Technical Support. Africa: A Guide to Identification of Tick Species; Bioscience Reports: Edinburgh, UK, 2003. [Google Scholar]
- Wang’ang’a Oundo, J.; Villinger, J.; Jeneby, M.; Ong’amo, G.; Otiende, M.Y.; Makhulu, E.E.; Musa, A.A.; Ouso, D.O.; Wambua, L. Pathogens, Endosymbionts, and Blood-Meal Sources of Host-Seeking Ticks in the Fast-Changing Maasai Mara Wildlife Ecosystem. PLoS ONE 2020, 15, e0228366. [Google Scholar] [CrossRef]
- Chiuya, T.; Masiga, D.K.; Falzon, L.C.; Bastos, A.D.S.; Fèvre, E.M.; Villinger, J. Tick-Borne Pathogens, Including Crime-an-Congo Haemorrhagic Fever Virus, at Livestock Markets and Slaughterhouses in Western Kenya. Transbound. Emerg. Dis. 2020, 68, 2429–2445. [Google Scholar] [CrossRef]
- Getange, D.; Bargul, J.L.; Kanduma, E.; Collins, M.; Bodha, B.; Denge, D.; Chiuya, T.; Githaka, N.; Younan, M.; Fèvre, E.M.; et al. Ticks and Tick-Borne Pathogens Associated with Dromedary Camels (Camelus dromedarius) in Northern Kenya. Microorganisms 2021, 9, 1414. [Google Scholar] [CrossRef]
- Endoh, D.; Mizutani, T.; Kirisawa, R.; Maki, Y.; Saito, H.; Kon, Y.; Morikawa, S.; Hayashi, M. Species-Independent Detection of RNA Virus by Representational Difference Analysis Using Non-Ribosomal Hexanucleotides for Reverse Transcription. Nucleic Acids Res. 2005, 33, e65. [Google Scholar] [CrossRef] [PubMed]
- Junglen, S.; Kopp, A.; Kurth, A.; Pauli, G.; Ellerbrok, H.; Leendertz, F.H. A New Flavivirus and a New Vector: Characterization of a Novel Flavivirus Isolated from Uranotaenia Mosquitoes from a Tropical Rain Forest. J. Virol. 2009, 83, 4462–4468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marklewitz, M.; Dutari, L.C.; Paraskevopoulou, S.; Page, R.A.; Loaiza, J.R.; Junglen, S. Diverse Novel Phleboviruses in Sandflies from the Panama Canal Area, Central Panama. J. Gen. Virol. 2019, 100, 938–949. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 2014, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A Novel Method for Rapid Multiple Sequence Alignment Based on Fast Fourier Transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Jones, P.; Binns, D.; Chang, H.Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-Scale Protein Function Classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [Green Version]
- R: The R Project for Statistical Computing. Available online: https://www.r-project.org/ (accessed on 4 November 2021).
- Christopher, J.; Williams, C.M.M. A Critique of Methods of Sampling and Reporting Pathogens in Populations of Fish. J. Aquat. Anim. Health 2001, 13, 300–309. [Google Scholar]
- Cowling, D.W.; Gardner, I.A.; Johnson, W.O. Comparison of Methods for Estimation of Individual-Level Prevalence Based on Pooled Samples. Prev. Vet. Med. 1999, 39, 211–225. [Google Scholar] [CrossRef]
- Sergeant, E.S.G. Epitools Epidemiological Calculators. Available online: https://epitools.ausvet.com.au/ (accessed on 4 November 2021).
- Lv, J.; Wu, S.; Zhang, Y.; Chen, Y.; Feng, C.; Yuan, X.; Jia, G.; Deng, J.; Wang, C.; Wang, Q.; et al. Assessment of Four DNA Fragments (COI, 16S RDNA, ITS2, 12S RDNA) for Species Identification of the Ixodida (Acari: Ixodida). Parasites Vectors 2014, 7, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sameroff, S.; Tokarz, R.; Charles, R.A.; Jain, K.; Oleynik, A.; Che, X.; Georges, K.; Carrington, C.V.; Lipkin, W.I.; Oura, C. Viral Diversity of Tick Species Parasitizing Cattle and Dogs in Trinidad and Tobago. Sci. Rep. 2019, 9, 10421. [Google Scholar] [CrossRef] [Green Version]
- De Souza, W.M.; Fumagalli, M.J.; Torres Carrasco, A.D.O.; Romeiro, M.F.; Modha, S.; Seki, M.C.; Gheller, J.M.; Daffre, S.; Nunes, M.R.T.; Murcia, P.R.; et al. Viral Diversity of Rhipicephalus Microplus Parasitizing Cattle in Southern Brazil. Sci. Rep. 2018, 8, 16315. [Google Scholar] [CrossRef] [PubMed]
- Dinçer, E.; Hacioǧlu, S.; Kar, S.; Emanet, N.; Brinkmann, A.; Nitsche, A.; Özkul, A.; Linton, Y.M.; Ergünay, K. Survey and Characterization of Jingmen Tick Virus Variants. Viruses 2019, 11, 71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailey, A.; Coulson, I.M. Habitat Association of Tortoises Geochelone Pardalis and Kinixys Spekii in Sengwa Wildlife Research Area, Zimbabwe. J. Herpetol. 1995, 5, 305–309. [Google Scholar]
- Demaya, G.S.; Benansio, J.S.; Petrozzi, F.; Di Vittorio, M.; Dendi, D.; John, E.F.; Luiselli, L. Habitat Use and Spatial Niche Overlap of Sympatric Savannah Tortoises at Multiple Spatial Scales in South Sudan. J. Arid Environ. 2020, 183, 104287. [Google Scholar] [CrossRef]
- Marschang, R.E. Viruses Infecting Reptiles. Viruses 2011, 3, 2087–2126. [Google Scholar] [CrossRef] [Green Version]
- Allender, M.C.; Abd-Eldaim, M.; Schumacher, J.; Mcruer, D.; Christian, L.S.; Kennedy, M. PCR Prevalence of Ranavirus in Free-Ranging Eastern Box Turtles (Terrapene carolina carolina) at Rehabilitation Centers in Three Southeastern US States. J. Wildl. Dis. 2011, 47, 759–764. [Google Scholar] [CrossRef]
- Marschang, R.E.; Kolesnik, E.; Mittenzwei, F. Detection of Testudinid Herpesvirus Type 4 in a Leopard Tortoise (Stigmochelys pardalis). Tierärztl Prax. 2016, 44, 283–286. [Google Scholar] [CrossRef]
- Omondi, D.; Masiga, D.K.; Fielding, B.C.; Kariuki, E.; Ajamma, Y.U.; Mwamuye, M.M.; Ouso, D.O.; Villinger, J. Molecular Detection of Tick-Borne Pathogen Diversities in Ticks from Livestock and Reptiles along the Shores and Adjacent Islands of Lake Victoria and Lake Baringo, Kenya. Front. Vet. Sci. 2017, 4, 73. [Google Scholar] [CrossRef] [Green Version]
- Ergönül, Ö. Crimean-Congo Haemorrhagic Fever. Lancet Infect. Dis. 2006, 6, 203–214. [Google Scholar] [CrossRef]
- Spengler, J.R.; Estrada-Pena, A.; Garrison, A.R.; Schmaljohn, C.; Spiropoulou, C.F.; Bergeron, E.; Bente, D. A Chronological Review of Experimental Infection Studies of the Role of Wild Animals and Livestock in the Maintenance and Transmission of Crimean-Congo Hemorrhagic Fever Virus. Antiviral Res. 2016, 135, 31–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pietzsch, M.; Quest, R.; Hillyard, P.D.; Medlock, J.M.; Leach, S. Importation of Exotic Ticks into the United Kingdom via the International Trade in Reptiles. Exp. Appl. Acarol. 2006, 38, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, D.; Kuwata, R.; Kimura, T.; Shimoda, H.; Fujita, R.; Faizah, A.N.; Kai, I.; Matsumura, R.; Kuroda, Y.; Watanabe, S.; et al. Detection of Jingmenviruses in Japan with Evidence of Vertical Transmission in Ticks. Viruses 2021, 13, 2547. [Google Scholar] [CrossRef]
- Kholodilov, I.S.; Belova, O.A.; Morozkin, E.S.; Litov, A.G.; Ivannikova, A.Y.; Makenov, M.T.; Shchetinin, A.M.; Aibulatov, S.V.; Bazarova, G.K.; Bell-Sakyi, L.; et al. Geographical and Tick-Dependent Distribution of Flavi-Like Alongshan and Yanggou Tick Viruses in Russia. Viruses 2021, 13, 458. [Google Scholar] [CrossRef]
- Wang, Z.D.; Wang, W.; Wang, N.N.; Qiu, K.; Zhang, X.; Tana, G.; Liu, Q.; Zhu, X.Q. Prevalence of the Emerging Novel Alongshan Virus Infection in Sheep and Cattle in Inner Mongolia, Northeastern China. Parasites Vectors 2019, 12, 450. [Google Scholar] [CrossRef] [Green Version]
- Ferron, F.; Rancurel, C.; Longhi, S.; Cambillau, C.; Henrissat, B.; Canard, B. VaZyMolO: A Tool to Define and Classify Modularity in Viral Proteins. J. Gen. Virol. 2005, 86, 743–749. [Google Scholar] [CrossRef]
- Brahma, R.K.; Dixit, V.; Sangwan, A.K.; Doley, R. Identification and Characterization of Rhipicephalus (Boophilus) microplus and Haemaphysalis bispinosa Ticks (Acari: Ixodidae) of Northeast India by ITS2 and 16S RDNA Sequences and Morphological Analysis. Exp. Appl. Acarol. 2014, 62, 253–265. [Google Scholar] [CrossRef]
- Chitimia, L.; Lin, R.Q.; Cosoroaba, I.; Braila, P.; Song, H.Q.; Zhu, X.Q. Molecular Characterization of Hard and Soft Ticks from Romania by Sequences of the Internal Transcribed Spacers of Ribosomal DNA. Parasitol. Res. 2009, 105, 907–911. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| County | Sampling Site | Tick Species | Goats (n) | Sheep (n) | Tortoises (n) | Cattle (n) |
|---|---|---|---|---|---|---|
| Kajiado | Oloisinyai | Rh. appendiculatus | 0 (0/4) | 0.5 (1/30) | 0 (0/0) | 0 (0/5) |
| Baringo | Ntepes | Rh. appendiculatus | 0 (0/84) | 4.4 (16/55) | 0 (0/0) | 0 (0/0) |
| Sandai | Rh. evertsi evertsi | 0 (0/0) | 42.3 (1/2) | 0 (0/0) | 0 (0/2) | |
| Rh. appendiculatus | 0.5 (1/28) | 0 (0/7) | 0 (0/0) | 0 (0/20) | ||
| Hy. truncatum | 0 (0/2) | 29.3 (1/2) | 0 (0/0) | 0 (0/3) | ||
| Logumgum | Hy. truncatum | 15.5 (1/3) | 0 (0/2) | 0 (0/0) | 0 (0/0) | |
| Rh. appendiculatus | 1.5 (4/35) | 3.3 (7/30) | 0 (0/0) | 0 (0/0) | ||
| Kapkuikui | Am. sp. | 0 (0/0) | 0 (0/0) | 77.1 (27/35) | 0 (0/0) | |
| Am. sparsum | 0 (0/0) | 0 (0/0) | 85.7 (6/7) | 0 (0/0) | ||
| Am. nuttalli | 0 (0/0) | 0 (0/0) | 40.0 (2/5) | 0 (0/0) | ||
| Total (n) | 0.3 (6/156) | 1.8 (26/128) | 74.5 (35/47) | 0 (0/30) |
| Code | County | Sampling Site | Time of Sample Collection | Species | Pool Size | Host | Sequence Length (nt) | GenBank Accession No |
|---|---|---|---|---|---|---|---|---|
| MT146 | Baringo | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 594 | ON158858 |
| MT147 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 595 | ON158857 | |
| MT148 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 602 | ON158854 | |
| MT149 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 594 | ON158864 | |
| MT150 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 591 | ON158852 | |
| MT151 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 589 | ON158862 | |
| MT152 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 593 | ON158853 | |
| MT153 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 586 | ON158856 | |
| MT154 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 604 | ON158860 | |
| MT155 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 588 | ON158859 | |
| MT156 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 591 | ON158851 | |
| MT157 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 591 | ON158855 | |
| MT158 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 590 | ON158865 | |
| MT159 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 595 | ON158861 | |
| MT160 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 596 | ON158863 | |
| MT161 | Ntepes | 10 August 2019 | Rh. appendiculatus | 8♀ | Sheep | 592 | ON158850 | |
| MT290 | Kapkuikui | 10 August 2019 | Am. sparsum | 1♂ | Tortoise | 577 | ON158844 | |
| MT291 | Kapkuikui | 10 August 2019 | Am. sparsum | 1♂ | Tortoise | 578 | ON158847 | |
| MT292 | Kapkuikui | 10 August 2019 | Am. sparsum | 1♂ | Tortoise | 580 | ON158831 | |
| MT293 | Kapkuikui | 10 August 2019 | Am. sparsum | 1♂ | Tortoise | 561 | ON158846 | |
| MT294 | Kapkuikui | 10 August 2019 | Am. sparsum | 1♂ | Tortoise | 590 | ON158837 | |
| MT295 | Kapkuikui | 10 August 2019 | Am. sparsum | 1♂ | Tortoise | 539 | ON158845 | |
| MT297 | Kapkuikui | 10 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 559 | ON158835 | |
| MT298 | Kapkuikui | 10 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 538 | ON158841 | |
| MT299 | Kapkuikui | 10 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 532 | ON158822 | |
| MT300 | Kapkuikui | 10 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 531 | ON158842 | |
| MT302 | Kapkuikui | 12 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 580 | ON158834 | |
| MT303 | Kapkuikui | 12 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 559 | ON158840 | |
| MT304 | Kapkuikui | 12 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 559 | ON158839 | |
| MT305 | Kapkuikui | 12 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 489 | ON158843 | |
| MT306 | Kapkuikui | 12 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 524 | ON158836 | |
| MT307 | Kapkuikui | 12 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 516 | ON158849 | |
| MT308 | Kapkuikui | 12 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 520 | ON158838 | |
| MT309 | Kapkuikui | 12 August 2019 | Am. nuttalli | 1♀ | Tortoise | 562 | ON158866 | |
| MT314 | Kapkuikui | 12 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 577 | ON158829 | |
| MT317 | Kapkuikui | 12 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 578 | ON158833 | |
| MT319 | Kapkuikui | 12 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 562 | ON158827 | |
| MT320 | Kapkuikui | 13 August 2019 | Am. nuttalli | 1♂ | Tortoise | 603 | ON158832 | |
| MT323 | Kapkuikui | 13 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 588 | ON158821 | |
| MT324 | Kapkuikui | 13August 2019 | Amblyomma sp. | 1♂ | Tortoise | 584 | ON158830 | |
| MT327 | Kapkuikui | 14 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 531 | ON158848 | |
| MT328 | Kapkuikui | 14 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 520 | ON158820 | |
| MT329 | Kapkuikui | 14 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 531 | ON158867 | |
| MT330 | Kapkuikui | 15 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 514 | ON158826 | |
| MT331 | Kapkuikui | 15 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 519 | ON158823 | |
| MT332 | Kapkuikui | 15 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 565 | ON158824 | |
| MT333 | Kapkuikui | 15 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 568 | ON158825 | |
| MT334 | Kapkuikui | 15 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 575 | ON158819 | |
| MT335 | Kapkuikui | 15 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 575 | ON158818 | |
| MT336 | Kapkuikui | 15 August 2019 | Amblyomma sp. | 1♂ | Tortoise | 571 | ON158828 | |
| MT4 | Logumgum | 15 August 2019 | Rh. appendiculatus | 8♂ | Sheep | ‡ | ‡ | |
| MT8 | Logumgum | 15 August 2019 | Rh. appendiculatus | 8♂ | Sheep | ‡ | ‡ | |
| MT19 | Logumgum | 15 August 2019 | Rh. appendiculatus | 8♀ | Sheep | ‡ | ‡ | |
| MT23 | Logumgum | 15 August 2019 | Rh. appendiculatus | 8♀ | Sheep | ‡ | ‡ | |
| MT26 | Logumgum | 15 August 2019 | Rh. appendiculatus | 8♀ | Sheep | ‡ | ‡ | |
| MT29 | Logumgum | 15 August 2019 | Rh. appendiculatus | 8♀ | Sheep | ‡ | ‡ | |
| MT31 | Logumgum | 15 August 2019 | Rh. appendiculatus | 8♀ | Sheep | ‡ | ‡ | |
| MT42 | Logumgum | 15 August 2019 | Rh. appendiculatus | 8♀ | Goat | ‡ | ‡ | |
| MT54 | Logumgum | 15 August 2019 | Rh. appendiculatus | 8♂ | Goat | ‡ | ‡ | |
| MT55 | Logumgum | 15 August 2019 | Rh. appendiculatus | 8♂ | Goat | ‡ | ‡ | |
| MT61 | Logumgum | 15 August 2019 | Hy. truncatum | 2♂ | Goat | ‡ | ‡ | |
| MT62 | Logumgum | 15 August 2019 | Rh. appendiculatus | 8♀ | Goat | ‡ | ‡ | |
| MT101 | Sandai | 15 August 2019 | Rh. appendiculatus | 8♂ | Goat | ‡ | ‡ | |
| MT136 | Sandai | 15 August 2019 | Hy. truncatum | 2♂ | Sheep | ‡ | ‡ | |
| MT144 | Sandai | 15 August 2019 | Rh. evertsi evertsi | 2♂ | Sheep | ‡ | ‡ | |
| MT313 | Kapkuikui | 12 Auguast 2019 | Amblyomma sp. | 1♂ | Tortoise | ‡ | ‡ | |
| T3 * | Kapkuikui | 29 September 2019 | 1♀ | 596 | ON158817 | |||
| T2 * | Kapkuikui | 29 September 2019 | 1♀ | ‡ | ‡ | |||
| KT125 | Kajiado | Oloisinyai | 19 July 2020 | Rh. appendiculatus | 8♂ | Sheep | ‡ | ‡ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogola, E.O.; Kopp, A.; Bastos, A.D.S.; Slothouwer, I.; Marklewitz, M.; Omoga, D.; Rotich, G.; Getugi, C.; Sang, R.; Torto, B.; et al. Jingmen Tick Virus in Ticks from Kenya. Viruses 2022, 14, 1041. https://doi.org/10.3390/v14051041
Ogola EO, Kopp A, Bastos ADS, Slothouwer I, Marklewitz M, Omoga D, Rotich G, Getugi C, Sang R, Torto B, et al. Jingmen Tick Virus in Ticks from Kenya. Viruses. 2022; 14(5):1041. https://doi.org/10.3390/v14051041
Chicago/Turabian StyleOgola, Edwin O., Anne Kopp, Armanda D. S. Bastos, Inga Slothouwer, Marco Marklewitz, Dorcus Omoga, Gilbert Rotich, Caroline Getugi, Rosemary Sang, Baldwyn Torto, and et al. 2022. "Jingmen Tick Virus in Ticks from Kenya" Viruses 14, no. 5: 1041. https://doi.org/10.3390/v14051041
APA StyleOgola, E. O., Kopp, A., Bastos, A. D. S., Slothouwer, I., Marklewitz, M., Omoga, D., Rotich, G., Getugi, C., Sang, R., Torto, B., Junglen, S., & Tchouassi, D. P. (2022). Jingmen Tick Virus in Ticks from Kenya. Viruses, 14(5), 1041. https://doi.org/10.3390/v14051041