
SARS-CoV-2 Variants in Paraguay: Detection and Surveillance with an Economical and Scalable Molecular Protocol

 ,
,  ,
,  , , , ,
, , , ,
Abstract
:1. Introduction
2. Methods
2.1. Clinical Samples
2.2. Spike SNP Design and Performance
2.3. Sequencing
2.4. Statistics
3. Results
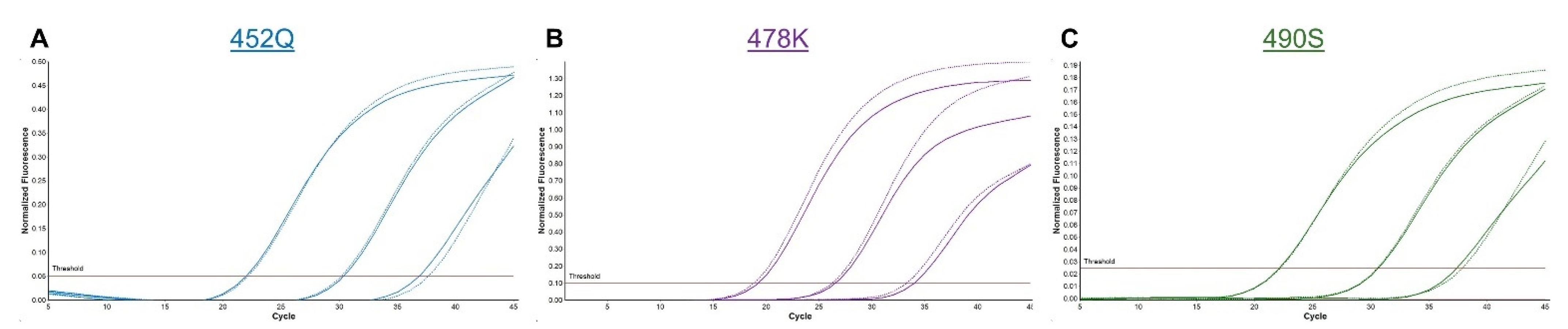
3.1. Spike SNP Optimization
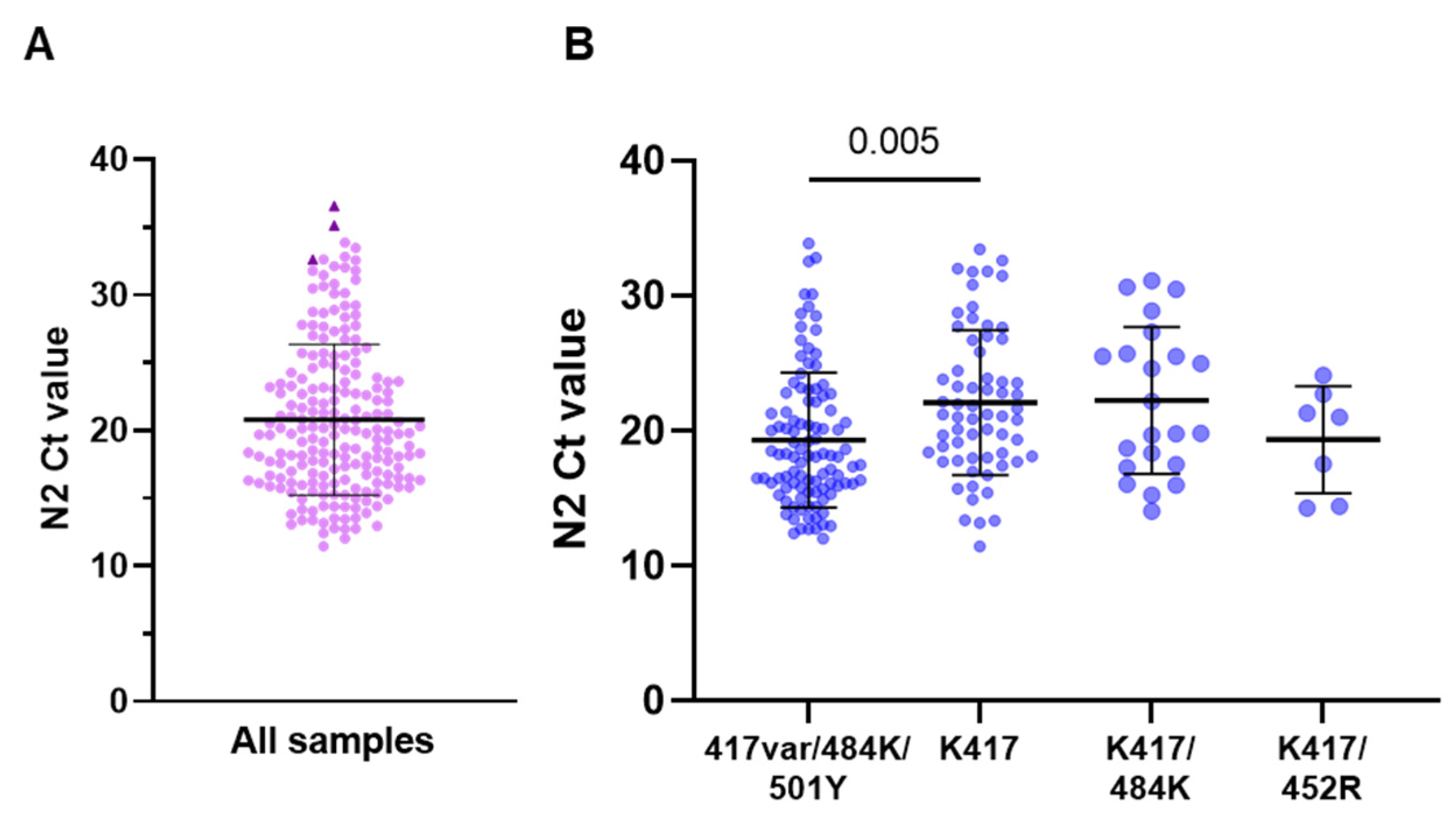
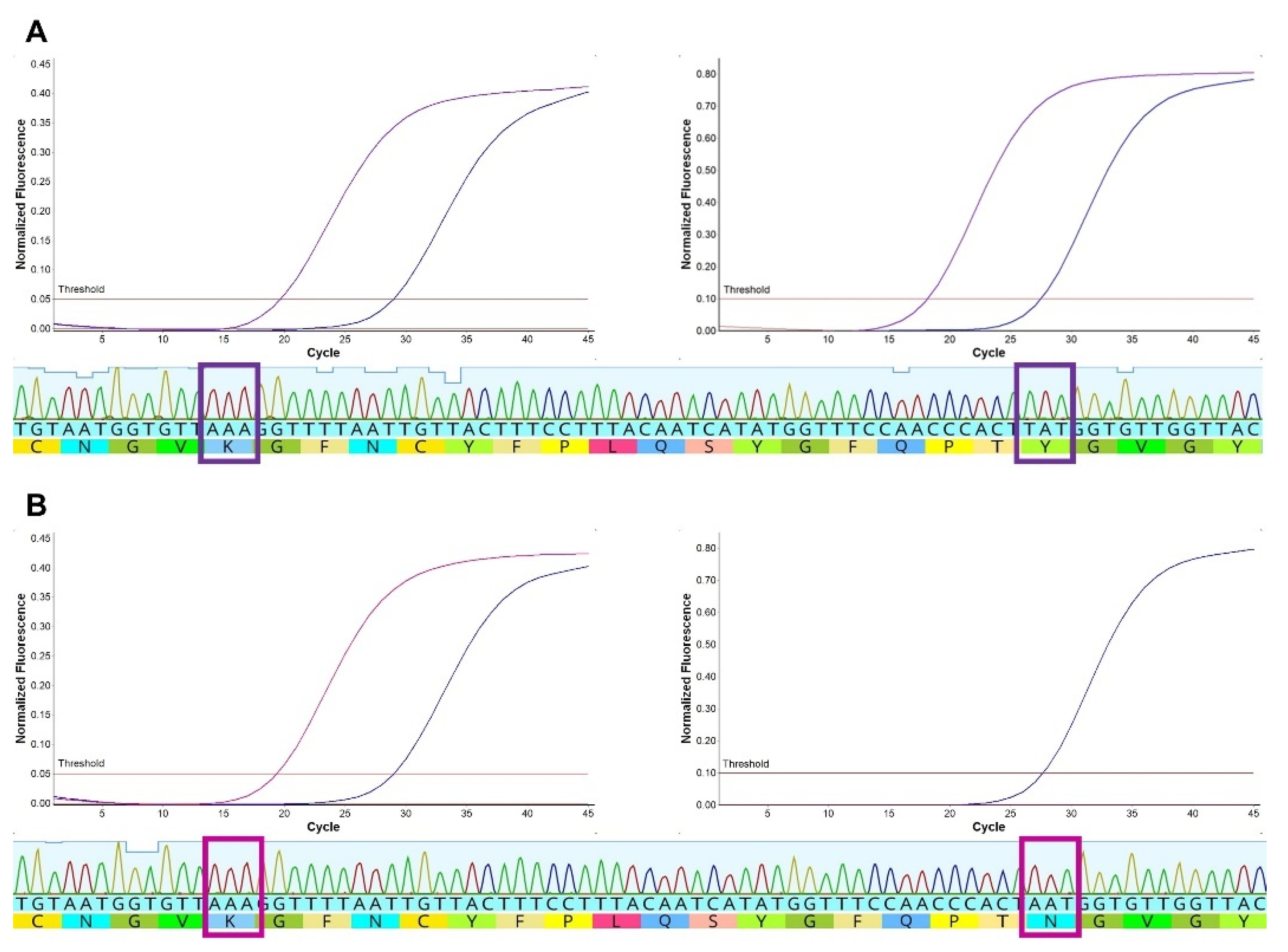
3.2. SNP Detection in Clinical Samples
3.3. Amplicon and Whole-Genome Sequencing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Centers for Disease Control and Prevention SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html#Concern (accessed on 2 September 2021).
- Gardy, J.L.; Loman, N.J. Towards a genomics-informed, real-time, global pathogen surveillance system. Nat. Rev. Genet. 2018, 19, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Vavrek, D.; Speroni, L.; Curnow, K.J.; Oberholzer, M.; Moeder, V.; Febbo, P.G. Genomic surveillance at scale is required to detect newly emerging strains at an early timepoint. medRxiv 2021. [Google Scholar] [CrossRef]
- Pan American Health Organization. Recommendations for Reporting and Notification of Sars-Cov-2 Variants of Concern and Variants of Interest; Pan American Health Organization: Washington, WA, USA, 2021. [Google Scholar]
- World Health Organization. Guidance for Surveillance of Sars-Cov-2 Variants; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Banada, P.; Green, R.; Banik, S.; Chopoorian, A.; Streck, D.; Jones, R.; Chakravorty, S.; Alland, D. A simple RT-PCR melting temperature assay to rapidly screen for widely circulating SARS-CoV-2 variants. J. Clin. Microbiol. 2021, 59, e00845-21. [Google Scholar] [CrossRef] [PubMed]
- Bedotto, M.; Fournier, P.E.; Houhamdi, L.; Levasseur, A.; Delerce, J.; Pinault, L.; Padane, A.; Chamieh, A.; Tissot-Dupont, H.; Brouqui, P.; et al. Implementation of an in-house real-time reverse transcription-PCR assay for the rapid detection of the SARS-CoV-2 Marseille-4 variant. J. Clin. Virol. 2021, 139, 104814. [Google Scholar] [CrossRef]
- Blairon, L.; Cupaiolo, R.; Piteus, S.; Beukinga, I.; Tre-Hardy, M. The challenge of screening SARS-CoV-2 variants of concern with RT-qPCR: One variant can hide another. J. Virol. Methods 2021, 297, 114248. [Google Scholar] [CrossRef]
- Camp, J.V.; Buchta, C.; Jovanovic, J.; Puchhammer-Stockl, E.; Benka, B.; Griesmacher, A.; Aberle, S.W.; Goerzer, I. RT-PCR based SARS-CoV-2 variant screening assays require careful quality control. J. Clin. Virol. 2021, 141, 104905. [Google Scholar] [CrossRef]
- Heijnen, L.; Elsinga, G.; de Graaf, M.; Molenkamp, R.; Koopmans, M.P.G.; Medema, G. Droplet digital RT-PCR to detect SARS-CoV-2 signature mutations of variants of concern in wastewater. Sci. Total Environ. 2021, 799, 149456. [Google Scholar] [CrossRef]
- La Rosa, G.; Mancini, P.; Bonanno Ferraro, G.; Veneri, C.; Iaconelli, M.; Lucentini, L.; Bonadonna, L.; Brusaferro, S.; Brandtner, D.; Fasanella, A.; et al. Rapid screening for SARS-CoV-2 variants of concern in clinical and environmental samples using nested RT-PCR assays targeting key mutations of the spike protein. Water Res. 2021, 197, 117104. [Google Scholar] [CrossRef]
- Norz, D.; Grunwald, M.; Olearo, F.; Fischer, N.; Aepfelbacher, M.; Pfefferle, S.; Lutgehetmann, M. Evaluation of a fully automated high-throughput SARS-CoV-2 multiplex qPCR assay with built-in screening functionality for del-HV69/70- and N501Y variants such as B.1.1.7. J. Clin. Virol. 2021, 141, 104894. [Google Scholar] [CrossRef]
- Tierling, S.; Kattler, K.; Vogelgesang, M.; Pfuhl, T.; Lohse, S.; Lo Porto, C.; Schmitt, B.; Nastasja, S.; Salhab, A.; Smola, S.; et al. Rapid base-specific calling of SARS-CoV-2 variants of concern using combined RT-PCR melting curve screening and SIRPH technology. Open Forum Infect. Dis. 2021, 8, ofab364. [Google Scholar] [CrossRef]
- Vega-Magana, N.; Sanchez-Sanchez, R.; Hernandez-Bello, J.; Venancio-Landeros, A.A.; Pena-Rodriguez, M.; Vega-Zepeda, R.A.; Galindo-Ornelas, B.; Diaz-Sanchez, M.; Garcia-Chagollan, M.; Macedo-Ojeda, G.; et al. RT-qPCR assays for rapid detection of the N501Y, 69–70del, K417N, and E484K SARS-CoV-2 mutations: A screening strategy to identify variants with clinical impact. Front. Cell. Infect. Microbiol. 2021, 11, 672562. [Google Scholar] [CrossRef]
- Vogels, C.B.F.; Breban, M.I.; Ott, I.M.; Alpert, T.; Petrone, M.E.; Watkins, A.E.; Kalinich, C.C.; Earnest, R.; Rothman, J.E.; Goes de Jesus, J.; et al. Multiplex qPCR discriminates variants of concern to enhance global surveillance of SARS-CoV-2. PLoS Biol. 2021, 19, e3001236. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Jean, S.; Eltringham, R.; Madison, J.; Snyder, P.; Tu, H.; Jones, D.M.; Leber, A.L. Mutation-specific SARS-CoV-2 PCR screen: Rapid and accurate detection of variants of concern and the identification of a newly emerging variant with spike L452R mutation. J. Clin. Microbiol. 2021, 59, e0092621. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Miller, J.A.; Verghese, M.; Sibai, M.; Solis, D.; Mfuh, K.O.; Jiang, B.; Iwai, N.; Mar, M.; Huang, C.; et al. Multiplex SARS-CoV-2 genotyping reverse transcriptase PCR for population-level variant screening and epidemiologic surveillance. J. Clin. Microbiol. 2021, 59, e0085921. [Google Scholar] [CrossRef] [PubMed]
- Yaniv, K.; Ozer, E.; Shagan, M.; Lakkakula, S.; Plotkin, N.; Bhandarkar, N.S.; Kushmaro, A. Direct RT-qPCR assay for SARS-CoV-2 variants of concern (Alpha, B.1.1.7 and Beta, B.1.351) detection and quantification in wastewater. Environ. Res. 2021, 201, 111653. [Google Scholar] [CrossRef]
- Zelyas, N.; Pabbaraju, K.; Croxen, M.A.; Lynch, T.; Buss, E.; Murphy, S.A.; Shokoples, S.; Wong, A.; Kanji, J.N.; Tipples, G. Precision response to the rise of the SARS-CoV-2 B.1.1.7 variant of concern by combining novel PCR assays and genome sequencing for rapid variant detection and surveillance. Microbiol. Spectr. 2021, 9, e0031521. [Google Scholar] [CrossRef]
- Babiker, A.; Immergluck, K.; Stampfer, S.D.; Rao, A.; Bassit, L.; Su, M.; Nguyen, V.; Stittleburg, V.; Ingersoll, J.M.; Bradley, H.L.; et al. Single-amplicon, multiplex real-time RT-PCR with tiled probes to detect SARS-CoV-2 spike mutations associated with variants of concern. J. Clin. Microbiol. 2021, 59, e014462. [Google Scholar] [CrossRef]
- Ministerio de Salud Pública y Bienestar Social. Primer Caso del Nuevo Coronavirus en El Paraguay. Available online: https://www.mspbs.gov.py/portal/20535/primer-caso-del-nuevo-coronavirus-en-el-paraguay.html (accessed on 1 September 2021).
- Ministerio de Salud Pública y Bienestar Social. Available online: https://www.mspbs.gov.py/index.php (accessed on 25 August 2021).
- Sobarzo, P.; Rolón López, J.C.; Narváez Serra, P.F.; López Cañete, S.A. Clinical characteristics of the first 60 patients with SARS CoV-2 admitted to the National Hospital for the period June–August 2020. Rev. Virtual Soc. Parag. Med. Int. 2021, 8, 69–77. [Google Scholar] [CrossRef]
- Vargas-Correa, A.; Mereles, E.F.; Segovia, C.N.; Gimenez, A.A.; Santacruz, L.; Ojeda, M.L.; Kunzle, E.H.; Samudio, M. Clinical-epidemiological characteristics of patients confirmed with COVID-19 from the Department of Alto Paraná, Paraguay. Rev. Salud Pública Parag. 2021, 11, 54–61. [Google Scholar] [CrossRef]
- Brinkmann, A.; Ulm, S.L.; Uddin, S.; Forster, S.; Seifert, D.; Oehme, R.; Corty, M.; Schaade, L.; Michel, J.; Nitsche, A. AmpliCoV: Rapid whole-genome sequencing using multiplex PCR amplification and real-time Oxford nanopore MinION sequencing enables rapid variant identification of SARS-CoV-2. Front. Microbiol. 2021, 12, 651151. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.W.; Bleicker, T.; Brunink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eur. Surveill. Bull. Eur. Mal. Transm. Eur. Commun. Dis. Bull. 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waggoner, J.J.; Stittleburg, V.; Pond, R.; Saklawi, Y.; Sahoo, M.K.; Babiker, A.; Hussaini, L.; Kraft, C.S.; Pinsky, B.A.; Anderson, E.J.; et al. Triplex real-time RT-PCR for severe acute respiratory syndrome coronavirus 2. Emerg. Infect. Dis. 2020, 26, 1633–1635. [Google Scholar] [CrossRef]
- Quick, J. nCoV-2019 Sequencing Protocol v3 (LoCost) V.3. Available online: https://www.protocols.io/view/ncov-2019-sequencing-protocol-v3-locost-bh42j8ye (accessed on 1 February 2021).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; O’Toole, A.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and epidemiology of the P.1 SARS-CoV-2 lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef] [PubMed]
- Naveca, F.G.; Nascimento, V.; de Souza, V.C.; Corado, A.L.; Nascimento, F.; Silva, G.; Costa, A.; Duarte, D.; Pessoa, K.; Mejia, M.; et al. COVID-19 in Amazonas, Brazil, was driven by the persistence of endemic lineages and P.1 emergence. Nat. Med. 2021, 27, 1230–1238. [Google Scholar] [CrossRef]
- Singh, J.; Rahman, S.A.; Ehtesham, N.Z.; Hira, S.; Hasnain, S.E. SARS-CoV-2 variants of concern are emerging in India. Nat. Med. 2021, 27, 1131–1133. [Google Scholar] [CrossRef]
- Liu, Z.; VanBlargan, L.A.; Bloyet, L.M.; Rothlauf, P.W.; Chen, R.E.; Stumpf, S.; Zhao, H.; Errico, J.M.; Theel, E.S.; Liebeskind, M.J.; et al. Identification of SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. Cell Host Microbe 2021, 29, 477–488. [Google Scholar] [CrossRef]
- Francisco, R.D.S., Jr.; Benites, L.F.; Lamarca, A.P.; de Almeida, L.G.P.; Hansen, A.W.; Gularte, J.S.; Demoliner, M.; Gerber, A.L.; de C Guimarães, A.P.; Antunes, A.K.E.; et al. Pervasive transmission of E484K and emergence of VUI-NP13L with evidence of SARS-CoV-2 co-infection events by two different lineages in Rio Grande do Sul, Brazil. Virus Res. 2021, 296, 198345. [Google Scholar] [CrossRef]
- Lythgoe, K.A.; Hall, M.; Ferretti, L.; de Cesare, M.; MacIntyre-Cockett, G.; Trebes, A.; Andersson, M.; Otecko, N.; Wise, E.L.; Moore, N.; et al. SARS-CoV-2 within-host diversity and transmission. Science 2021, 372, eabg082. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence a | Concentration (nM) b | Location (5′–3′) c |
|---|---|---|---|
| Primers | |||
| SpikeSNP Ext d | TGAAGTCAGACAAATCGCTCC | 400 | 22,777–22,797 |
| SpikeSNP R1 d | TGGTGCATGTAGAAGTTCAAAAG | 400 | 23,103–23,125 |
| LNA Probes | |||
| K417 d | FAM-CAAACTGGA+A+A+G+ATTGCTG-3IABkFQ | 200 | 22,802–22,820 |
| 484K d | Cy5-ACCTTGTAATGGTGT+T+A+AAGGTTTT-3IAbRQSp | 200 | 22,996–23,020 |
| 501Y d | HEX-CCCAC+T+T+ATGGTGTTGG-3IABkFQ | 200 | 23,057–23,073 |
| Unmodified Probes | |||
| 452R d | CFR610-ATAATTACCGGTATAGATTGTTTAGGAAGT-BHQ-2 | 200 | 22,908–22,937 |
| 484K | Q670-CACCTTGTAATGGTGTTAAAGGTTTTAA-BHQ-2 | 200 | 22,995–23,022 |
| 484Q | Q670-CACCTTGTAATGGTGTTCAAGGTTTTAA-BHQ-2 | 200 | 22,995–23,022 |
| 501Y | CFO560-CAACCCACTTATGGTGTTGGTTAC-BHQ-1 | 200 | 23,054–23,077 |
| 452Q | CFR610-ATAATTACCAGTATAGATTGTTTAGGAAGT-BHQ-2 | 200 | 22,908–22,937 |
| 490S | FAM-ACTCTCCTTTACAATCATATGGTTTCC-BHQ-1 | 200 | 23,028–23,054 |
| 478K (original) | CFO560-CGGTAGCAAACCTTGTAATGGTG-BHQ-1 | 200 | 22,987–23,009 |
| 478K (revised) | CFO560-CGGTARCAAACCTTGTAATGGTG-BHQ-1 | 200 | 22,987–23,009 |
| Category | n (%) | Age, Years, Mean (SD) a | Day of Symptoms, Mean (SD) b | Ct, Mean (SD) c | Sequencing Confirmed, n/N (%) d |
|---|---|---|---|---|---|
| N2 detected | 201 (100) | 42.6 (16.4) | 4.8 (3.0) | 20.8 (5.6) | ─ |
| Spike SNP detected | 198 (98.5) | 42.5 (16.4) | 4.8 (3.0) | 20.6 (5.3) | 181/181 (100) |
| Genotype e | |||||
| 417var/484K/501Y | 102 (51.5) | 42.8 (14.9) | 4.5 (2.4) | 19.3 (5.0) | 96/96 (100) |
| K417 | 64 (32.3) | 42.6 (18.8) | 5.1 (3.7) | 22.1 (5.4) | 56/56 (100) |
| K417/484K | 22 (11.1) | 37.4 (14.2) | 4.8 (2.7) | 22.2 (5.4) | 20/20 (100) f |
| K417/452R | 7 (3.5) | 54.0 (15.8) | 5.0 (4.6) | 19.4 (4.0) | 7/7 (100) |
| K417/490S | 1 (0.5) | 41 | 4 | 15.2 | 1/1 (100) |
| K417/501Y | 1 (0.5) | 68 | 5 | 19.7 | 1/1 (100) |
| K417/484K/501Y | 1 (0.5) | 19 | 10 | 32.1 | NA |
| Sample Code | Spike SNP Genotype | Nanopore Sequencing | |||
|---|---|---|---|---|---|
| GISAID Accession No. | Nextstrain CladeWHO Label | Pango Lineage | Spike Deletions/Amino Acid Substitutions a | ||
| PY21-100 | 417var/484K/501Y | EPI_ISL_4071892 | 20J (V3) gamma | P.1 | L18F/T20N/P26S/D138Y/R190S/K417T/E484K/N501Y/D614G/H655Y/T1027I/V1176F |
| PY21-9 | 417var/484K/501Y | EPI_ISL_2444778 | 20J (V3) gamma | P.1 | L18F//T20N/P26S/D138Y/R190S/K417T/E484K/N501Y/D614G/H655Y/T1027I/V1176F |
| PY21-57 | 417var/484K/501Y | EPI_ISL_4137476 | 20J (V3) gamma | P.1 | L18F/T20N/P26S/D138Y/R190S/K417T/E484K/N501Y/D614G/H655Y/T1027I/V1176F |
| PY21-58 | 417var/484K/501Y | EPI_ISL_4071897 | 20J (V3) gamma | P.1 | L18F/T20N/P26S7D138Y/R190S/K417T/E484K/N501Y/D614G/H655Y/T1027I/V1176F |
| PY21-84 | 417var/484K/501Y | EPI_ISL_4071898 | 20J (V3) gamma | P.1 | L18F/T20N/P26S/D138Y/R190S/K417T/E484K/N501Y/D614G/H655Y/T1027I/V1176F |
| PY21-16 | 417var/484K/501Y | EPI_ISL_4071893 | 20J (V3) gamma | P.1 | L18F/T20N/P26S/D138Y/R190S/K417T/E484K/N501Y/D614G/H655Y/T1027I/V1176F |
| PY21-11 | 417var/484K/501Y | EPI_ISL_4071900 | 20J (V3) gamma | P.1 | L18F/T20N/P26S/D138Y/L189F/R190S/K417T/E484K/N501Y/D614G/H655Y/T1027I/V1176F |
| PY21-90 | 417var/484K/501Y | EPI_ISL_4071899 | 20J (V3) gamma | P.1.2 | L18F/T20N/P26S/T95I/D138Y/R190S/K417T/E484K/N501Y/D614G/H655Y/Q675H/T1027I/V1176F/V1228L |
| PY21-150 | K417 | EPI_ISL_2234899 | 20B | B.1.1.33 | D614G |
| PY21-153 | K417 | EPI_ISL_2234891 | 20B | B.1.1.33 | D614G |
| PY21-140 | K417 | EPI_ISL_2234880 | 20B | B.1.1.28 | D614G/V1176F |
| PY21-145 | K417 | EPI_ISL_4084605 | 20B | B.1.1.28 | D614G/V1176F |
| PY21-146 | K417 | EPI_ISL_2234881 | 20B | B.1.1.28 | D614G/V1176F |
| PY21-98 | K417 | EPI_ISL_2444788 | 20B | B.1.1.28 | D614G/V1176F |
| PY21-152 | K417 | EPI_ISL_2234885 | 20B | B.1.1.28 | T22I/D614G/V1176F |
| PY21-144 | K417 | EPI_ISL_4071901 | 20B | B.1.1.28 | D614G/N1135Y/V1176F |
| PY21-43 | K417 | EPI_ISL_4071895 | 20B | B.1.1.28 | A575S/D614G/V1176F |
| PY21-44 | K417 | EPI_ISL_4084604 | 20B | B.1.1.28 | T572I/D614G/V1176F |
| PY21-47 | K417 | EPI_ISL_4071894 | 20B | B.1.1.28 | A575S/D614G/V1176F |
| PY21-148 | K417 | EPI_ISL_2234894 | 20B | B.1.1.28 | V6F/L18F/D614G/V1176F |
| PY21-147 | K417 | EPI_ISL_2234887 | 20B | B.1.1.28 | Q14H/D614G/D1153A/V1176F |
| PY21-154 | K417 | EPI_ISL_2234893 | 20B | B.1.1.277 | D614G/V1176F |
| PY21-52 | K417/484K | EPI_ISL_2444780 | 20B (zeta) | P.2 | E484K/D614G/V1176F |
| PY21-157 | K417/484K | EPI_ISL_2234879 | 20B (zeta) | P.2 | L5F/E484K/D614G/A771S/V1176F |
| PY21-143 | K417 | EPI_ISL_2234889 | 20B | N.3 | T76I/D614G |
| PY21-141 | K417 | EPI_ISL_2234883 | 20C | B.1.499 | T478R/D614G |
| PY21-149 | K417 | EPI_ISL_2234897 | 20C | B.1.499 | T478R/D614G |
| PY21-102 | K417/452R | EPI_ISL_4071896 | 19B | A.2.5.2 | L452R/D614G |
| PY21-156 | K417/501Y | EPI_ISL_4084606 | 20I (V1) alpha | B.1.1.7 | DEL69-70/DEL144N501Y/A570D/D614G/P681H/T716I/S982A/D1118H |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martinez, M.; Nguyen, P.-V.; Su, M.; Cardozo, F.; Valenzuela, A.; Franco, L.; Galeano, M.E.; Rojas, L.E.; Díaz Acosta, C.C.; Fernández, J.; et al. SARS-CoV-2 Variants in Paraguay: Detection and Surveillance with an Economical and Scalable Molecular Protocol. Viruses 2022, 14, 873. https://doi.org/10.3390/v14050873
Martinez M, Nguyen P-V, Su M, Cardozo F, Valenzuela A, Franco L, Galeano ME, Rojas LE, Díaz Acosta CC, Fernández J, et al. SARS-CoV-2 Variants in Paraguay: Detection and Surveillance with an Economical and Scalable Molecular Protocol. Viruses. 2022; 14(5):873. https://doi.org/10.3390/v14050873
Chicago/Turabian StyleMartinez, Magaly, Phuong-Vi Nguyen, Maxwell Su, Fátima Cardozo, Adriana Valenzuela, Laura Franco, María Eugenia Galeano, Leticia Elizabeth Rojas, Chyntia Carolina Díaz Acosta, Jonás Fernández, and et al. 2022. "SARS-CoV-2 Variants in Paraguay: Detection and Surveillance with an Economical and Scalable Molecular Protocol" Viruses 14, no. 5: 873. https://doi.org/10.3390/v14050873
APA StyleMartinez, M., Nguyen, P. -V., Su, M., Cardozo, F., Valenzuela, A., Franco, L., Galeano, M. E., Rojas, L. E., Díaz Acosta, C. C., Fernández, J., Ortiz, J., del Puerto, F., Mendoza, L., Nara, E., Rojas, A., & Waggoner, J. J. (2022). SARS-CoV-2 Variants in Paraguay: Detection and Surveillance with an Economical and Scalable Molecular Protocol. Viruses, 14(5), 873. https://doi.org/10.3390/v14050873