
Bayesian Phylodynamic Analysis Reveals the Dispersal Patterns of African Swine Fever Virus

 , ,
, , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Sequence Alignment of ASFV
2.2. Annotation of ASFV Genomes
2.3. Detection of Homologous Recombination
2.4. Phylogenetic Analysis
2.5. Time Signal Detection and Evolutionary Mutation Rate
2.6. Population Dynamic
2.7. Genetic Differentiation among Different ASFV Populations
2.8. Phylogeographic Analysis
3. Results
3.1. ASFV Genome Sequences
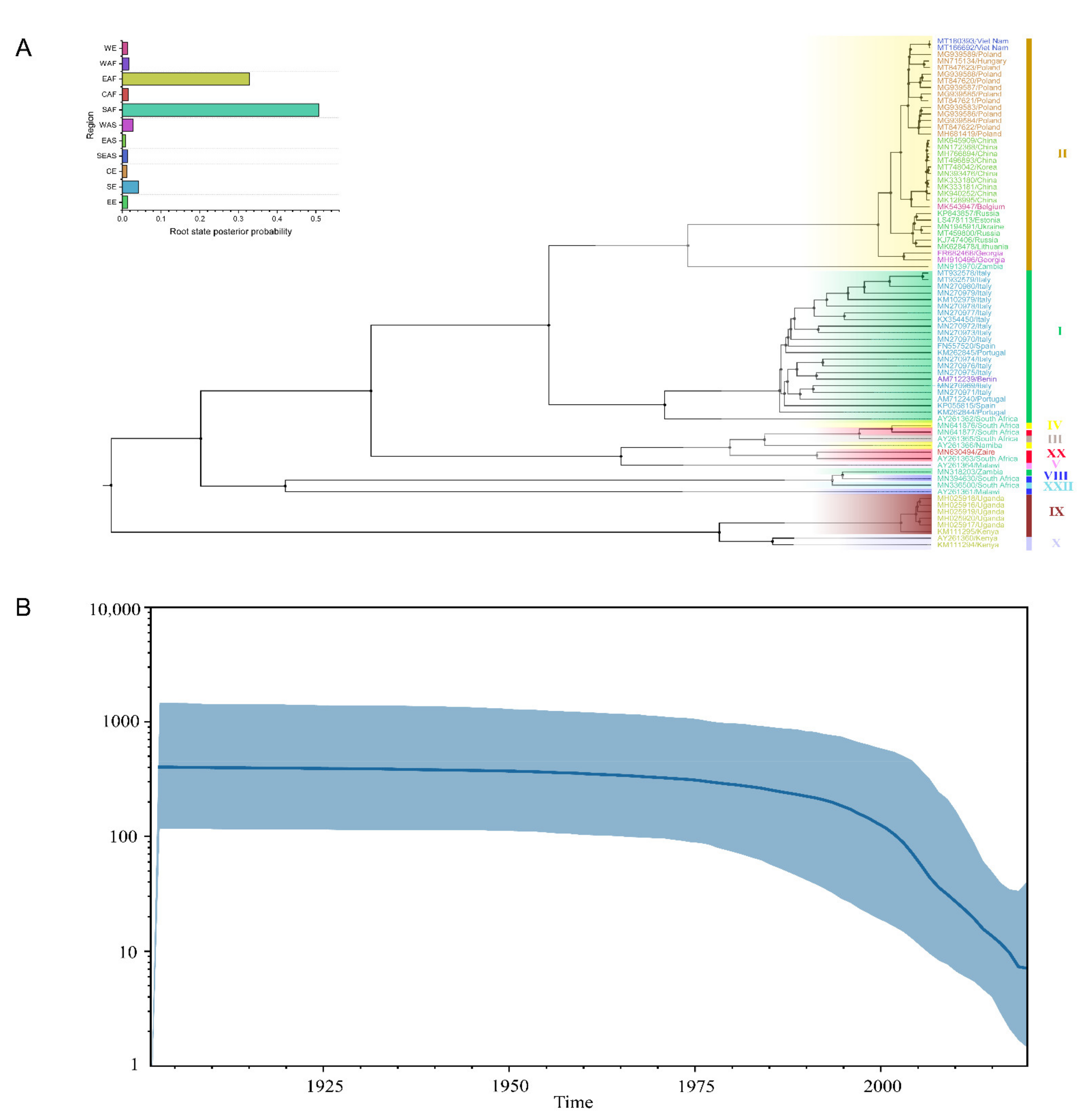
3.2. Phylogenetic Inference
3.3. Evolutionary Mutation Rate and Bayesian Phylodynamic Analysis
3.4. Migration of ASFV in the World
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, N.; Zhao, D.; Wang, J.; Zhang, Y.; Wang, M.; Gao, Y.; Li, F.; Wang, J.; Bu, Z.; Rao, Z.; et al. Architecture of African swine fever virus and implications for viral assembly. Science 2019, 366, 640–644. [Google Scholar] [CrossRef] [PubMed]
- Quembo, C.J.; Jori, F.; Vosloo, W.; Heath, L. Genetic characterization of African swine fever virus isolates from soft ticks at the wildlife/domestic interface in Mozambique and identification of a novel genotype. Transbound. Emerg. Dis. 2018, 65, 420–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, C.; Miskin, J.; Hernáez, B.; Fernandez-Zapatero, P.; Soto, L.; Cantó, C.; Rodríguez-Crespo, I.; Dixon, L.; Escribano, J.M. African swine fever virus protein p54 interacts with the microtubular motor complex through direct binding to light-chain dynein. J. Virol. 2001, 75, 9819–9827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez, J.M.; García-Escudero, R.; Salas, M.L.; Andrés, G. African swine fever virus structural protein p54 is essential for the recruitment of envelope precursors to assembly sites. J. Virol. 2004, 78, 4299–4313. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Han, D.; Zhang, Y.; Zhang, K.; Du, N.; Tong, W.; Li, G.; Zheng, H.; Liu, C.; Gao, F.; et al. Identification of one novel epitope targeting p54 protein of African swine fever virus using monoclonal antibody and development of a capable ELISA. Res. Vet. Sci. 2021, 141, 19–25. [Google Scholar] [CrossRef]
- Minoungou, G.L.; Diop, M.; Dakouo, M.; Ouattara, A.K.; Settypalli, T.B.K.; Lo, M.M.; Sidibe, S.; Kanyala, E.; Kone, Y.S.; Diallo, M.S.; et al. Molecular characterization of African Swine fever viruses in Burkina Faso, Mali, and Senegal 1989-2016: Genetic diversity of ASFV in West Africa. Transbound. Emerg. Dis. 2021, 68, 2842–2852. [Google Scholar] [CrossRef]
- Patrick, B.N.; Machuka, E.M.; Githae, D.; Banswe, G.; Amimo, J.O.; Ongus, J.R.; Masembe, C.; Bishop, R.P.; Steinaa, L.; Djikeng, A.; et al. Evidence for the presence of African swine fever virus in apparently healthy pigs in South-Kivu Province of the Democratic Republic of Congo. Vet. Microbiol. 2020, 240, 108521. [Google Scholar] [CrossRef]
- Molini, U.; Mushonga, B.; Settypalli, T.B.K.; Dundon, W.G.; Khaiseb, S.; Jago, M.; Cattoli, G.; Lamien, C.E. Molecular characterization of African swine fever virus from outbreaks in Namibia in 2018. Transbound. Emerg. Dis. 2020, 67, 1008–1014. [Google Scholar] [CrossRef]
- Montgomery, R.E. On a form of swine fever occurring in British East Africa (Kenya Colony). J. Comparat. Pathol. Ther. 1921, 34, 159–191. [Google Scholar] [CrossRef] [Green Version]
- Revilla, Y.; Pérez-Núñez, D.; Richt, J.A. African Swine Fever Virus Biology and Vaccine Approaches. Adv. Virus Res. 2018, 100, 41–74. [Google Scholar]
- Costard, S.; Mur, L.; Lubroth, J.; Sanchez-Vizcaino, J.M.; Pfeiffer, D.U. Epidemiology of African swine fever virus. Virus Res. 2013, 173, 191–197. [Google Scholar] [CrossRef]
- Gallardo, M.C.; Reoyo, A.T.; Fernández-Pinero, J.; Iglesias, I.; Muñoz, M.J.; Arias, M.L. African swine fever: A global view of the current challenge. Porcine Health Manag. 2015, 1, 21. [Google Scholar] [CrossRef] [Green Version]
- Rowlands, R.J.; Michaud, V.; Heath, L.; Hutchings, G.; Oura, C.; Vosloo, W.; Dwarka, R.; Onashvili, T.; Albina, E.; Dixon, L.K. African swine fever virus isolate, Georgia, 2007. Emerg. Infect. Dis. 2008, 14, 1870–1874. [Google Scholar] [CrossRef]
- Smietanka, K.; Wozniakowski, G.; Kozak, E.; Niemczuk, K.; Fraczyk, M.; Bocian, L.; Kowalczyk, A.; Pejsak, Z. African Swine Fever Epidemic, Poland, 2014–2015. Emerg. Infect. Dis. 2016, 22, 1201–1207. [Google Scholar] [CrossRef] [Green Version]
- Kolbasov, D.; Titov, I.; Tsybanov, S.; Gogin, A.; Malogolovkin, A. African Swine Fever Virus, Siberia, Russia, 2017. Emerg. Infect. Dis. 2018, 24, 796–798. [Google Scholar] [CrossRef]
- Linden, A.; Licoppe, A.; Volpe, R.; Paternostre, J.; Lesenfants, C.; Cassart, D.; Garigliany, M.; Tignon, M.; van den Berg, T.; Desmecht, D.; et al. Summer 2018: African swine fever virus hits north-western Europe. Transbound. Emerg. Dis. 2019, 66, 54–55. [Google Scholar] [CrossRef]
- Garigliany, M.; Desmecht, D.; Tignon, M.; Cassart, D.; Lesenfant, C.; Paternostre, J.; Volpe, R.; Cay, A.B.; van den Berg, T.; Linden, A. Phylogeographic Analysis of African Swine Fever Virus, Western Europe, 2018. Emerg. Infect. Dis. 2019, 25, 184–186. [Google Scholar] [CrossRef]
- Gonzales, W.; Moreno, C.; Duran, U.; Henao, N.; Bencosme, M.; Lora, P.; Reyes, R.; Núñez, R.; De Gracia, A.; Perez, A.M. African swine fever in the Dominican Republic. Transbound. Emerg. Dis. 2021, 68, 3018–3019. [Google Scholar] [CrossRef]
- Zhou, X.; Li, N.; Luo, Y.; Liu, Y.; Miao, F.; Chen, T.; Zhang, S.; Cao, P.; Li, X.; Tian, K.; et al. Emergence of African Swine Fever in China, 2018. Transbound. Emerg. Dis. 2018, 65, 1482–1484. [Google Scholar] [CrossRef] [Green Version]
- Bao, J.; Wang, Q.; Lin, P.; Liu, C.; Li, L.; Wu, X.; Chi, T.; Xu, T.; Ge, S.; Liu, Y.; et al. Genome comparison of African swine fever virus China/2018/AnhuiXCGQ strain and related European p72 Genotype II strains. Transbound. Emerg. Dis. 2019, 66, 1167–1176. [Google Scholar] [CrossRef]
- Xiong, D.; Zhang, X.; Xiong, J.; Yu, J.; Wei, H. Rapid genome-wide sequence typing of African swine fever virus based on alleles. Virus Res. 2021, 297, 198357. [Google Scholar] [CrossRef] [PubMed]
- EFSA Panel on Animal Health and Welfare. Scientific opinion on African swine fever. EFSA J. 2014, 12, 3628. [Google Scholar]
- European Food Safety Authority; Boklund, A.; Cay, B.; Depner, K.; Foldi, Z.; Guberti, V.; Masiulis, M.; Miteva, A.; More, S.; Olsevskis, E.; et al. Epidemiological analyses of African swine fever in the European Union (November 2017 until November 2018). EFSA J. 2018, 16, e05494. [Google Scholar] [PubMed]
- Zani, L.; Dietze, K.; Dimova, Z.; Forth, J.H.; Denev, D.; Depner, K.; Alexandrov, T. African Swine Fever in a Bulgarian Backyard Farm-A Case Report. Vet. Sci. 2019, 6, 94. [Google Scholar] [CrossRef] [Green Version]
- Nurmoja, I.; Mõtus, K.; Kristian, M.; Niine, T.; Schulz, K.; Depner, K.; Viltrop, A. Epidemiological analysis of the 2015–2017 African swine fever outbreaks in Estonia. Prev. Vet. Med. 2020, 181, 104556. [Google Scholar] [CrossRef]
- Chastagner, A.; Pereira de Oliveira, R.; Hutet, E.; Le Dimna, M.; Paboeuf, F.; Lucas, P.; Blanchard, Y.; Dixon, L.; Vial, L.; Le Potier, M.F. Coding-Complete Genome Sequence of an African Swine Fever Virus Strain Liv13/33 Isolate from Experimental Transmission between Pigs and Ornithodoros moubata Ticks. Microbiol. Resour. Announc. 2020, 9, e00185-20. [Google Scholar] [CrossRef]
- Tcherepanov, V.; Ehlers, A.; Upton, C. Genome Annotation Transfer Utility (GATU): Rapid annotation of viral genomes using a closely related reference genome. BMC Genom. 2006, 7, 150. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega for making accurate alignments of many protein sequences. Protein Sci. 2018, 27, 135–145. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Huson, D.H. SplitsTree: Analyzing and visualizing evolutionary data. Bioinformatics 1998, 14, 68–73. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef] [Green Version]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Duchêne, S.; Duchêne, D.; Holmes, E.C.; Ho, S.Y. The Performance of the Date-Randomization Test in Phylogenetic Analyses of Time-Structured Virus Data. Mol. Biol. Evol. 2015, 32, 1895–1906. [Google Scholar] [CrossRef] [Green Version]
- Rieux, A.; Khatchikian, C.E. tipdatingbeast: An r package to assist the implementation of phylogenetic tip-dating tests using beast. Mol. Ecol. Resour. 2017, 17, 608–613. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Patrono, L.V.; Pleh, K.; Samuni, L.; Ulrich, M.; Rothemeier, C.; Sachse, A.; Muschter, S.; Nitsche, A.; Couacy-Hymann, E.; Boesch, C.; et al. Monkeypox virus emergence in wild chimpanzees reveals distinct clinical outcomes and viral diversity. Nat. Microbiol. 2020, 5, 955–965. [Google Scholar] [CrossRef]
- Düx, A.; Lequime, S.; Patrono, L.V.; Vrancken, B.; Boral, S.; Gogarten, J.F.; Hilbig, A.; Horst, D.; Merkel, K.; Prepoint, B.; et al. Measles virus and rinderpest virus divergence dated to the sixth century BCE. Science 2020, 368, 1367–1370. [Google Scholar] [CrossRef]
- To, T.H.; Jung, M.; Lycett, S.; Gascuel, O. Fast Dating Using Least-Squares Criteria and Algorithms. Syst. Biol. 2016, 65, 82–97. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Minin, V.N.; Suchard, M.A. Counting labeled transitions in continuous-time Markov models of evolution. J. Math. Biol. 2008, 56, 391–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, L.; Song, J.; Liu, H.; Cai, J.; Lin, Q.; Xu, C.; Ding, C.; Liao, M.; Ren, T.; Xiang, B. Phylodynamic analyses of class I Newcastle disease virus isolated in China. Transboun. Emerg. Dis. 2021, 68, 1294–1304. [Google Scholar] [CrossRef] [PubMed]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [Green Version]
- Murray, G.G.; Wang, F.; Harrison, E.M.; Paterson, G.K.; Mather, A.E.; Harris, S.R.; Holmes, M.A.; Rambaut, A.; Welch, J.J. The effect of genetic structure on molecular dating and tests for temporal signal. Methods Ecol. Evol. 2016, 7, 80–89. [Google Scholar] [CrossRef]
- Wilkinson, P. The persistence of African swine fever in Africa and the Mediterranean. Prev. Vet. Med. 1984, 2, 71–82. [Google Scholar] [CrossRef]
- Motayo, B.O.; Oluwasemowo, O.O.; Olusola, B.A.; Opayele, A.V.; Faneye, A.O. Phylogeography and evolutionary analysis of African Rotavirus a genotype G12 reveals district genetic diversification within lineage III. Heliyon 2019, 5, e02680. [Google Scholar] [CrossRef] [Green Version]
- Simulundu, E.; Lubaba, C.H.; van Heerden, J.; Kajihara, M.; Mataa, L.; Chambaro, H.M.; Sinkala, Y.; Munjita, S.M.; Munang’andu, H.M.; Nalubamba, K.S.; et al. The Epidemiology of African Swine Fever in “Nonendemic” Regions of Zambia (1989–2015): Implications for Disease Prevention and Control. Viruses 2017, 9, 236. [Google Scholar] [CrossRef] [Green Version]
- Bisimwa, P.N.; Ongus, J.R.; Tiambo, C.K.; Machuka, E.M.; Bisimwa, E.B.; Steinaa, L.; Pelle, R. First detection of African swine fever (ASF) virus genotype X and serogroup 7 in symptomatic pigs in the Democratic Republic of Congo. Virol. J. 2020, 17, 135. [Google Scholar] [CrossRef]
- Bisimwa, P.N.; Ishara, L.K.; Wasso, D.S.; Bantuzeko, F.; Tonui, R.; Bwihangane, A.B. Detection and genetic characterization of African swine fever virus (ASFV) in clinically infected pigs in two districts in South Kivu province, Democratic Republic Congo. Heliyon 2021, 7, e06419. [Google Scholar] [CrossRef]
- Le, V.P.; Jeong, D.G.; Yoon, S.W.; Kwon, H.M.; Trinh, T.B.N.; Nguyen, T.L.; Bui, T.T.N.; Oh, J.; Kim, J.B.; Cheong, K.M.; et al. Outbreak of African Swine Fever, Vietnam, 2019. Emerg. Infect. Dis. 2019, 25, 1433–1435. [Google Scholar] [CrossRef]
- Gago, S.; Elena, S.F.; Flores, R.; Sanjuán, R. Extremely high mutation rate of a hammerhead viroid. Science 2009, 323, 1308. [Google Scholar] [CrossRef] [Green Version]
- Sanjuán, R. From molecular genetics to phylodynamics: Evolutionary relevance of mutation rates across viruses. PLoS Pathog. 2012, 8, e1002685. [Google Scholar] [CrossRef] [Green Version]
- Njau, E.P.; Domelevo Entfellner, J.B.; Machuka, E.M.; Bochere, E.N.; Cleaveland, S.; Shirima, G.M.; Kusiluka, L.J.; Upton, C.; Bishop, R.P.; Pelle, R.; et al. The first genotype II African swine fever virus isolated in Africa provides insight into the current Eurasian pandemic. Sci. Rep. 2021, 11, 13081. [Google Scholar] [CrossRef]
- Khomenko, S.; Beltrán-Alcrudo, D.; Rozstalnyy, A.; Gogin, A.; Kolbasov, D.; Pinto, J.; Lubroth, J.; Martin, V. African swine fever in the Russian Federation: Risk factors. Empres Watch 2013, 28, 1–14. [Google Scholar]
- Sánchez-Vizcaíno, J.M.; Mur, L.; Martínez-López, B. African swine fever (ASF): Five years around Europe. Vet. Microbiol. 2013, 165, 45–50. [Google Scholar] [CrossRef]
- Ge, S.; Li, J.; Fan, X.; Liu, F.; Li, L.; Wang, Q.; Ren, W.; Bao, J.; Liu, C.; Wang, H.; et al. Molecular Characterization of African Swine Fever Virus, China, 2018. Emerg. Infect. Dis. 2018, 24, 2131–2133. [Google Scholar] [CrossRef] [Green Version]
- Costard, S.; Jones, B.A.; Martínez-López, B.; Mur, L.; de la Torre, A.; Martínez, M.; Sánchez-Vizcaíno, F.; Sánchez-Vizcaíno, J.M.; Pfeiffer, D.U.; Wieland, B. Introduction of African swine fever into the European Union through illegal importation of pork and pork products. PLoS ONE 2013, 8, e61104. [Google Scholar] [CrossRef] [Green Version]
- Ramirez-Medina, E.; Vuono, E.A.; Pruitt, S.; Rai, A.; Espinoza, N.; Velazquez-Salinas, L.; Gladue, D.P.; Borca, M.V. Evaluation of an ASFV RNA Helicase Gene A859L for Virus Replication and Swine Virulence. Viruses 2021, 14, 10. [Google Scholar] [CrossRef]
- Ramirez-Medina, E.; Vuono, E.; Rai, A.; Pruitt, S.; Espinoza, N.; Velazquez-Salinas, L.; Pina-Pedrero, S.; Zhu, J.; Rodriguez, F.; Borca, M.V.; et al. Deletion of E184L, a Putative DIVA Target from the Pandemic Strain of African Swine Fever Virus, Produces a Reduction in Virulence and Protection against Virulent Challenge. J. Virol. 2022, 96, e0141921. [Google Scholar] [CrossRef]
- Gladue, D.P.; Ramirez-Medina, E.; Vuono, E.; Silva, E.; Rai, A.; Pruitt, S.; Espinoza, N.; Velazquez-Salinas, L.; Borca, M.V. Deletion of the A137R Gene from the Pandemic Strain of African Swine Fever Virus Attenuates the Strain and Offers Protection against the Virulent Pandemic Virus. J. Virol. 2021, 95, e0113921. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GenBank ID | Isolate | Host | Country | Population | Year | p72 | Length (bp) |
|---|---|---|---|---|---|---|---|
| Genotype | |||||||
| MN318203 | LIV_5_40 | Tick | Zambia | SAF | 1983 | I | 183,291 |
| MN630494 | Zaire | Pig | Zaire | CAF | 1977 | XX | 185,338 |
| MT166692 | ASFV_Hanoi_2019 | Pig | Viet Nam | SEAS | 2019 | II | 166,931 |
| MT180393 | ASFV_NgheAn_2019 | Pig | Viet Nam | SEAS | 2019 | II | 186,498 |
| MN194591 | ASFV/Kyiv/2016/131 | Pig | Ukraine | EE | 2016 | II | 191,911 |
| MH025919 | N10 | Pig | Uganda | EAF | 2015 | IX | 188,611 |
| MH025918 | R25 | Pig | Uganda | EAF | 2015 | IX | 188,630 |
| MH025920 | R35 | Pig | Uganda | EAF | 2015 | IX | 188,629 |
| MH025917 | R7 | Pig | Uganda | EAF | 2015 | IX | 188,628 |
| MH025916 | R8 | Pig | Uganda | EAF | 2015 | IX | 188,627 |
| KP055815 | BA71 | Pig | Spain | SE | 1971 | I | 180,365 |
| FN557520 | E75 | Pig | Spain | SE | 1975 | I | 181,187 |
| ASU18466 | BA71V | Vero cells | Spain | SE | 1971 | I | 170,101 |
| MT748042 | ASFV/Korea/pig/PaJu1/2019 | Pig | South Korea | EAS | 2019 | II | 190,597 |
| AY261362 | Mkuzi 1979 | Tick | South Africa | SAF | 1979 | I | 192,714 |
| MN394630 | SPEC_57 | Tick | South Africa | SAF | 1985 | VIII | 186,119 |
| AY261363 | Pretorisuskop/96/4 | Tick | South Africa | SAF | 1996 | XX | 190,324 |
| MN641876 | RSA_W1_1999 | Warthog | South Africa | SAF | 1999 | IV | 185,293 |
| MN641877 | RSA_2_2004 | Wild boar | South Africa | SAF | 2004 | XX | 188,502 |
| MN336500 | RSA_2_2008 | Tick | South Africa | SAF | 2008 | XXII | 187,866 |
| AY261365 | Warmbaths | Tick | South Africa | SAF | 1987 | III | 190,773 |
| KJ747406 | Kashino 04/13 | Wild boar | Russia | EE | 2013 | II | 189,387 |
| KP843857 | Odintsovo_02/14 | Pig | Russia | EE | 2014 | II | 189,333 |
| MT459800 | ASFV/Kabardino-Balkaria 19/WB-964 | Wild boar | Russia | EE | 2019 | II | 189,252 |
| KM262844 | L60 | Pig | Portugal | SE | 1960 | I | 182,362 |
| KM262845 | NHV | Pig | Portugal | SE | 1968 | I | 172,051 |
| AM712240 | OURT 88/3 | Tick | Portugal | SE | 1988 | I | 171,719 |
| MH681419 | ASFV/POL/2015/Podlaskie | Wild boar | Poland | CE | 2015 | II | 189,394 |
| MG939584 | Pol16_20538_o9 | Pig | Poland | CE | 2016 | II | 189,399 |
| MG939585 | Pol16_20540_o10 | Pig | Poland | CE | 2016 | II | 189,405 |
| MG939586 | Pol16_29413_o23 | Pig | Poland | CE | 2016 | II | 189,393 |
| MG939583 | Pol16_20186_o7 | Pig | Poland | CE | 2016 | II | 189,401 |
| MG939587 | Pol17_03029_C201 | Pig | Poland | CE | 2017 | II | 189,405 |
| MG939588 | Pol17_04461_C210 | Pig | Poland | CE | 2017 | II | 189,401 |
| MG939589 | Pol17_05838_C220 | Pig | Poland | CE | 2017 | II | 189,393 |
| MT847620 | Pol17_55892_C754 | Pig | Poland | CE | 2017 | II | 189,414 |
| MT847621 | Pol18_28298_O111 | Pig | Poland | CE | 2018 | II | 189,409 |
| MT847623 | Pol19_53050_C1959/19 | Pig | Poland | CE | 2019 | II | 189,356 |
| MT847622 | Pol17_31177_O81 | Pig | Poland | CE | 2017 | II | 189,422 |
| AY261366 | Warthog | Warthog | Namibia | SAF | 1980 | IV | 186,528 |
| AY261364 | Tengani 62 | Pig | Malawi | SAF | 1962 | V | 185,689 |
| AY261361 | Malawi Lil-20/1 (1983) | Tick | Malawi | SAF | 1983 | VIII | 187,612 |
| MK628478 | ASFV/LT14/1490 | Wild boar | Lithuania | EE | 2014 | II | 189,399 |
| AY261360 | Kenya 1950 | Pig | Kenya | EAF | 1950 | X | 193,886 |
| KM111294 | Ken05/Tk1 | Tick | Kenya | EAF | 2005 | X | 191,058 |
| KM111295 | Ken06.Bus | Pig | Kenya | EAF | 2006 | IX | 184,368 |
| MN270969 | 56/Ca/1978 | Pig | Italy | SE | 1978 | I | 183,636 |
| MN270970 | 57/Ca/1979 | Pig | Italy | SE | 1979 | I | 183,639 |
| MN270971 | 139/Nu/1981 | Pig | Italy | SE | 1981 | I | 183,645 |
| MN270972 | 140/Or/1985 | Pig | Italy | SE | 1985 | I | 183,723 |
| MN270973 | 85/Ca/1985 | Pig | Italy | SE | 1985 | I | 181,816 |
| MN270974 | 141/Nu/1990 | Pig | Italy | SE | 1990 | I | 183,720 |
| MN270975 | 142/Nu/1995 | Pig | Italy | SE | 1995 | I | 183,724 |
| MN270976 | 60/Nu/1997 | Pig | Italy | SE | 1997 | I | 181,651 |
| MN270977 | 26/Ss/2004 | Pig | Italy | SE | 2004 | I | 184,581 |
| MN270978 | 72407/Ss/2005 | Pig | Italy | SE | 2005 | I | 181,699 |
| KX354450 | 47/Ss/2008 | Pig | Italy | SE | 2008 | I | 184,638 |
| KM102979 | 26544/OG10 | Pig | Italy | SE | 2010 | I | 182,906 |
| MN270979 | 97/Ot/2012 | Pig | Italy | SE | 2012 | I | 184,206 |
| MN270980 | 22653/Ca/2014 | Pig | Italy | SE | 2014 | I | 181,869 |
| MT932578 | 103917/18 | Pig | Italy | SE | 2018 | I | 181,759 |
| MT932579 | 55234/18 | Pig | Italy | SE | 2018 | I | 181,761 |
| MN715134 | ASFV_HU_2018 | Wild boar | Hungary | CE | 2018 | II | 190,601 |
| FR682468 | Georgia 2007/1 | Pig | Georgia | WAS | 2007 | II | 189,344 |
| MH910496 | Georgia 2008/2 | Pig | Georgia | WAS | 2008 | II | 189,315 |
| MN913970 | Liv13/33 | Tick | Zambia | SAF | 1983 | I | 188,277 |
| LS478113 | Estonia 2014 | Wild boar | Estonia | EE | 2014 | II | 182,446 |
| MK645909 | ASFV-wbBS01 | Wild boar | China | EAS | 2018 | II | 189,394 |
| MK128995 | China/2018/AnhuiXCGQ | Pig | China | EAS | 2018 | II | 189,393 |
| MK333180 | Pig/HLJ/2018 | Pig | China | EAS | 2018 | II | 189,404 |
| MH766894 | ASFV-SY18 | Pig | China | EAS | 2018 | II | 189,354 |
| MK333181 | DB/LN/2018 | Pig | China | EAS | 2018 | II | 189,404 |
| MN172368 | ASFV/pig/China/CAS19-01/2019 | Pig | China | EAS | 2019 | II | 189,405 |
| MN393476 | ASFV Wuhan 2019-1 | Pig | China | EAS | 2019 | II | 190,576 |
| MN393477 | ASFV Wuhan 2019-2 | Pig | China | EAS | 2019 | II | 190,576 |
| MK940252 | CN/2019/InnerMongolia-AES01 | Wild boar | China | EAS | 2019 | II | 189,403 |
| MT496893 | GZ201801 | Pig | China | EAS | 2018 | II | 189,393 |
| AM712239 | Benin 97/1 | Pig | Benin | WAF | 1997 | I | 182,284 |
| MK543947 | Belgium/Etalle/wb/2018 | Wild boar | Belgium | WE | 2018 | II | 190,202 |
| Statistic | Observed Mean (95% CI) | Null Mean (95% CI) | p-Value |
|---|---|---|---|
| AI | 0.94 (0.55–1.32) | 6.86 (6.21–7.40) | 0 |
| PS | 12.48 (11.00–14.00) | 49.92 (47.24–52.10) | 0 |
| MC (EAF) | 8.00 (8.00–8.00) | 1.21 (1.01–1.96) | <0.01 |
| MC (SE) | 10.89 (6.00–18.00) | 2.06 (1.60–2.73) | <0.01 |
| MC (SAF) | 3.66 (3.00–4.00) | 1.38 (1.07–1.97) | <0.01 |
| MC (CAF) | 1.00 (1.00–1.00) | 1.00 (1.00–1.00) | >0.05 |
| MC (WAF) | 1.00 (1.00–1.00) | 1.00 (1.00–1.00) | >0.05 |
| MC (WAS) | 1.70 (1.00–2.00) | 1.00 (1.00–1.01) | <0.01 |
| MC (EE) | 2.55 (1.00–6.00) | 1.13 (1.00–1.57) | <0.05 & >0.01 |
| MC (CE) | 6.34 (3.00–12.00) | 1.56 (1.18–2.16) | <0.01 |
| MC (WE) | 1.00 (1.00–1.00) | 1.00 (1.00–1.03) | >0.05 |
| MC (EAS) | 7.22 (3.00–10.00) | 1.31 (1.04–2.00) | <0.01 |
| MC (SEAS) | 1.98 (2.00–2.00) | 1.00 (1.00–1.00) | <0.01 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Z.-J.; Jia, H.; Xie, C.-D.; Shagainar, J.; Feng, Z.; Zhang, X.; Li, K.; Zhou, R. Bayesian Phylodynamic Analysis Reveals the Dispersal Patterns of African Swine Fever Virus. Viruses 2022, 14, 889. https://doi.org/10.3390/v14050889
Shen Z-J, Jia H, Xie C-D, Shagainar J, Feng Z, Zhang X, Li K, Zhou R. Bayesian Phylodynamic Analysis Reveals the Dispersal Patterns of African Swine Fever Virus. Viruses. 2022; 14(5):889. https://doi.org/10.3390/v14050889
Chicago/Turabian StyleShen, Zhao-Ji, Hong Jia, Chun-Di Xie, Jurmt Shagainar, Zheng Feng, Xiaodong Zhang, Kui Li, and Rong Zhou. 2022. "Bayesian Phylodynamic Analysis Reveals the Dispersal Patterns of African Swine Fever Virus" Viruses 14, no. 5: 889. https://doi.org/10.3390/v14050889
APA StyleShen, Z. -J., Jia, H., Xie, C. -D., Shagainar, J., Feng, Z., Zhang, X., Li, K., & Zhou, R. (2022). Bayesian Phylodynamic Analysis Reveals the Dispersal Patterns of African Swine Fever Virus. Viruses, 14(5), 889. https://doi.org/10.3390/v14050889